Novel ABCB4 mutations in an infertile female with progressive familial intrahepatic cholestasis type 3:A case report
2022-03-18 02:16:52TianFuLiuJingJingHeLiangWangLingYiZhang
World Journal of Clinical Cases 2022年6期
lNTRODUCTlON
Progressive familial intrahepatic cholestasis(PFIC)comprises several rare,hereditary autosomalrecessive hepatic diseases that occur predominantly in neonates and infants.Intrahepatic cholestasis is the primary clinical sign observed in patients with PFIC[1].As the disease progresses,patients develop liver fibrosis,which progresses to cirrhosis and eventually hepatic failure[2].Based on the specific mutations in the gene,six types of PFIC have been defined,among which classical types 1,2 and 3 are more common[3].In terms of biochemical and histological characteristics,there are significant differences between PFIC3 and other types(Table 1).In particular,PFIC3 presents with elevated serum γ-glutamyl transpeptidase(GGT)[4].We found few reports of cases of PFIC3 in our search of the published literature,which included the last ten years.In particular,fertility in adult PFIC3 patients has not been reported.
CASE PRESENTATlON
Chief complaints
A 32-year-old female patient had experienced abnormal serum liver enzyme levels for 15 years.
History of present illness
Fifteen years previously,the patient was diagnosed with abnormal serum liver enzyme levels during the course of a high school physical examination.As she did not exhibit any symptoms of overall liver dysfunction,no action was taken at that time.Four years later,liver function index abnormalities were observed during an occupational health examination.The patient was admitted to a local hospital with no apparent cause of her abnormal serum enzyme levels.The patient was treated with some liverprotective drugs,but her liver function indices did not improve.The patient was not experiencing any adverse effects at that time and did not actively have her liver function indices monitored in subsequent years.One month prior to her admission to our hospital,the patient visited a maternal and child health hospital due to infertility,but routine examination on admission revealed significantly abnormal liver function indices.For further diagnosis and treatment,she was admitted to the Department of Hepatology at our hospital.
History of past illness
Thirteen years previously,the patient had undergone splenectomy due to a ruptured spleen caused by a car accident and recovered well.
Not a breath of wind moved, not a leaf stirred, all was silent as the grave, only on the still bosom of the lake thirty ducks, with brilliant plumage, swam about in the water
Personal and family history
During her ten years of marriage,the patient experienced continued infertility and did not take any infertility medications or oral contraceptives.One year previously,salpingography at a maternal and child health hospital showed partial obstruction of the fallopian tube,and the natural conception rate was low.The fertility tests of the patient’s spouse were normal.Therefore,fertilization(IVF)-assisted reproduction was recommended.However,due to financial reasons,the patient did not receive IVF.
The authors thank the patient for agreeing to report her case and for providing a detailed medical history.
New evidence demonstrates a spectrum of diseases resulting from heterozygous mutations in,ranging from the severe form seen in PFIC3 to milder,intermittent forms such as low-phospholipidassociated cholelithiasis syndrome(LPAC),ICP and adult-onset biliary fibrosis or cirrhosis[18-21].Previous studies have shown that reduced or absent transport of phosphatidylcholine is indeed associated with intrahepatic sludge or stone formation[22].The father of the patient had a genetic mutation in,including a 2950C>T;p.A984V mutation,and his preexisting intrahepatic bile duct stone may have been a phenotypic form of LPAC[23].Although the majority of cases of ICP present in the third trimester and are usually thought to be multifactorial in etiology,the patient's early pregnancy presentation may have been attributed to an underlying genetic susceptibility[24].gene defects are one of the major causes of biliary fibrosis,and Bernardo[25]reported that PFIC3-associated biliary fibrosis can be partially reversed after UDCA treatment.However,the coexistence ofvariants and infertility has not been reported in previously published literature.While not proof of causality,the mutations identified in our patient certainly suggest a possible susceptibility for her rare and unique presentation.
Three months later my friends and I gathered at the same restaurant. To life in the Big Apple! they cheered as we tapped our glasses together. My chance of a lifetime! We talked for hours. I told them of my plan to save money by moving out of my beach cottage and renting a room for the few remaining months. Our friend offered, I have a fellow South African friend who is considering renting one of the four bedrooms in his house. His name is Barry. A great guy. He scribbled6 on a napkin() . This is his number. He s a forty-two-year-old confirmed bachelor. Says he s much too busy being a single dad to be a husband.
Physical examination
Natural Science Foundation of Gansu Province,No.21JR7RA410.
Laboratory examinations
PFIC3 occurs rarely and is primarily a sporadic disease[5],and the incidence of PFIC3 in the general population has not been reported definitively.A previous assessment of disease occurrence demonstrated that PFIC3 incidence might be 1 in 500000 people[6].Patients exhibiting PFIC3 commonly develop cholestasis later in childhood and up to adolescence.Individuals with PFIC3 often exhibit recurring episodes of pruritus,jaundice,pale clay-like stools,hepatosplenomegaly,and gastrointestinal bleeding,which can progress to cirrhosis and liver failure before the onset of adulthood[7,8].Gastrointestinal bleeding that is associated with cirrhosis or the occurrence of portal hypertension is often the first presenting sign of the disorder in older children or young adults[9].
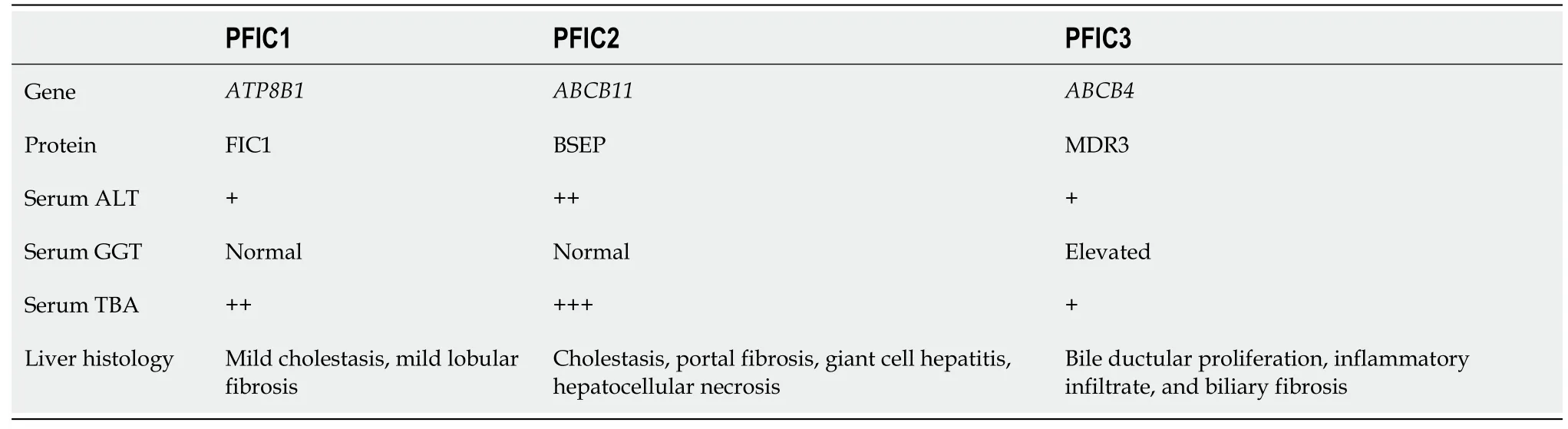
Imaging examinations
Abdominal magnetic resonance imaging(MRI)revealed an increased hepatic interstitium and multiple patchy abnormal signal shadows in hepatic segments VII and VIII.The presence of nodule regeneration was considered after enhanced examination using gadolinium-ethoxybenzyl-diethylenetriamine pentaacetic acid,which is a hepatocellular-specific contrast agent.No obstructions associated with the extrahepatic and intrahepatic bile ducts were observed following detailed examination using magnetic resonance cholangiopancreatography(Figure 1).Cardiac ultrasound and chest radiography showed no abnormalities.
Specialized examinations
We examined the sclera of the patient’s eyes for Kayser-Fleischer rings,but no rings were present.The transient elastography assessment for liver fibrosis was 14.6 kPa.
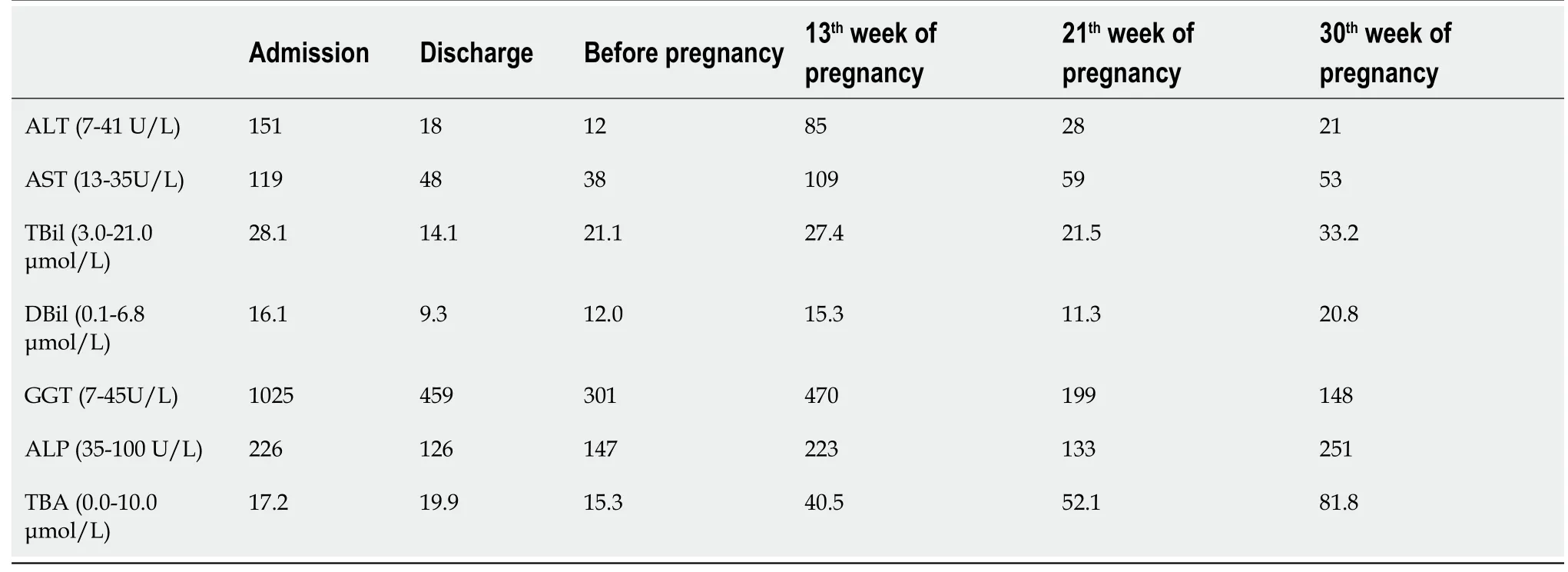
Pathological findings and immunohistochemical staining
With the consent of the patient,we performed an ultrasound-guided liver biopsy.Pathological examination revealed cholestatic liver fibrosis(modified Scheuer score S2).An analysis of histological samples revealed a significant decrease in multidrug-resistant protein 3(MDR3)protein staining compared to the levels of staining observed in samples from healthy subjects(Figure 2).
Gene mutational analysis
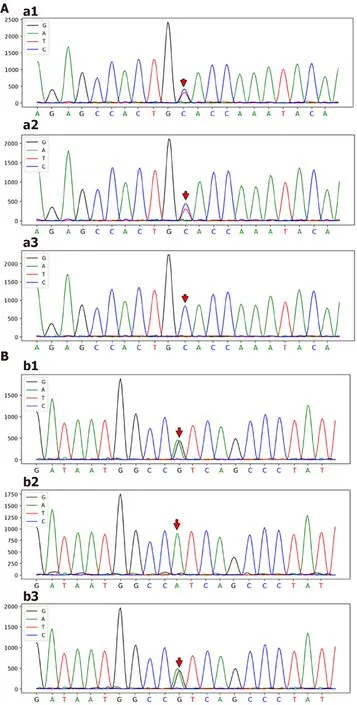
Genomic DNA was purified from peripheral blood samples.Based on Illumina NovaSeq6000 instruments and the xGensequencing Exome Research Panel,high-throughput sequencing was used to detect genomic mutations.The genetic testing results revealed two novel heterozygous mutations in thegene:a 2950C>T;p.A984V mutation(exon 24)and a 667A>G;p.I223V mutation(exon 7)(Figure 3).
FlNAL DlAGNOSlS
According to the above case data,the patient’s liver function,particularly the increase in serum GGT,was abnormal for many years.Immunohistochemical analysis of liver tissue showed that MDR3 staining was significantly decreased,and gene detection showed that there were two heterozygous mutations in thegene.The final diagnosis of the presented case was PFIC3.
TREATMENT
The patient received treatment with ursodeoxycholic acid(UDCA)750 mg/d.
OUTCOME AND FOLLOW-UP
When the patient was discharged,the GGT decreased significantly,and the other liver function indices were close to the normal values(Table 2).After consulting with family members and saving money,the patient went to the maternal and child health care hospital again,successfully became pregnantIVF,and stopped UDCA treatment on her own.Skin pruritus occurred at the 13th week of pregnancy,and serological examination showed that her liver function was significantly abnormal,in particular,TBA was higher than the initial value.The diagnosis was PFIC3 combined with intrahepatic cholestasis of pregnancy(ICP),and UDCA therapy was recommended.Her symptoms of itching were subsequently alleviated,and all liver function indices except TBA were improved.Fortunately,fetal indicators were normal.The liver function test results during the course of the disease are shown in Table 2.
DlSCUSSlON
Analysis of her laboratory data(November 3,2020)revealed the following results:total bilirubin,28.1 μmol/L;direct bilirubin,16.1 μmol/L;alanine aminotransferase(ALT),151 U/L;aspartate aminotransferase(AST),119 U/L;alkaline phosphatase(ALP),226 U/L;GGT,1025 U/L;total bile acid(TBA),17.2 μmol/L;and albumin,41.8 g/L(Table 2).Several markers for hepatitis viruses(hepatitis A,B,C,and E)were determined to be negative.The analyses for antibodies to cytomegalovirus antibodies and Epstein-Barr virus DNA were also negative.All autoimmune antibodies,including antimitochondrial and antinuclear lupus-related antibodies and Sjogren's syndrome-related antibodies were negative.We also assessed serum copper levels as well as ferritin and ceruloplasmin,and the levels were within normal ranges.Routine blood analysis,coagulation function,thyroid function,and several additional laboratory test results were found to be unremarkable.
In this case study,we analyzed a 32-year-old female Chinese patient who was asymptomatic.We used laboratory,MRI,and histological examinations to exclude other possible etiologies,including primary biliary cholangitis,Alagille syndrome,primary sclerosing cholangitis,Wilson's disease,and drug-induced liver injury.Subsequently,we highly suspected that this patient might have PFIC3 based on her high level of GGT.Therefore,we carried out a gene mutation analysis that revealed two novel heterozygous mutations in,namely,a 2950C>T;p.A984V mutation and a 667A>G;p.I223V mutation.Based on immunostaining for MDR3 in liver samples and genetic testing,the patient was diagnosed with PFIC3.
PFIC3 is caused by a mutation in thegene,which encodes MDR3[10].MDR3 is classified as a p-glycoprotein(pGp)and has been shown to be expressed in hepatocyte canalicular membranes[11].The MDR3 protein transfers phospholipids from hepatocytes into the bile ducts[12].Normally,phospholipids combine with bile salts that subsequently form microparticles.This process results in increased hydrophilicity and reduces the descaling effects produced by bile salts.These actions protect bile duct cells from toxic damage that can be induced by bile salts.A major function of phospholipids in the liver is to neutralize the detergent-like effects produced by hydrophobic bile salts[6,13].Thus,defects in the MDR3 protein can result in damage to the biliary epithelium and bile canaliculi,which ultimately can produce cholestasis.It should be noted that the primary defect that occurs with MDR3 deficiency does not lead to the retention of bile acids in hepatocytes,so cholestasis is not a direct result of the disorder[14].Symptoms,including cholestasis,develop as a result of the damage caused by the eventual cholangiopathy.Even patients who exhibit complete MDR3 deficiency may not present clinical symptoms for several years[15].When MDR3 function is only partially lost,patients commonly exhibit slow disease progression[16].Therefore,MDR3 deficiencies can result in a range of disease manifestations and ages at presentation.
Nearly 300variants that cause disease have been reported,of which approximately 50 are associated with PFIC3[7].The age at which the patient first exhibits signs of cholestatic disease,severity of the liver disorder and response to treatment have been shown to be correlated with the different mutations that occur in thegene[16].Patients with homozygous mutations tend to exhibit progressive intrahepatic cholestasis,which usually leads to liver failure in early childhood and requires liver transplantation[17].On the other hand,the age of disease onset in patients with heterozygous mutations is relatively high,and clinical symptoms are mild.
But the young King noticed that Trusty John always missed one door, and said: Why do you never open this one for me? There is something inside that would appal3 you, he answered
The patient indicated that she did not have a history of alcohol intake or the use of any hepatotoxic drugs.Her father had undergone surgery years previously to treat intrahepatic bile duct stones and recovered well from the surgery.Her mother,who died from a brain hemorrhage three years earlier,did not have liver disease in her medical history.Her brother’s liver function was normal.
Genetic sequencing of thegene in our patient revealed two novel heterozygous mutations,namely,a 2950C>T;p.A984V mutation and a 667A>G;p.I223V mutation.These two mutations were not included in the Human Gene Mutation Database or the ClinVar database.The effects on function resulting from the two mutations were evaluated using Polymorphism Phenotyping v2,Sorting Intolerant from Tolerant,and MutationTaster[26-28].The two novel mutations were predicted to have uncertain significance.Based on the familial identification,the locations of these two mutations were determined to be on different chromosomes,which resulted in a compound heterozygous mutation that might have resulted in a partial loss of function for MDR3.It is due to this compound heterozygous mutation that this patient has a rare and unique clinical phenotype.
The King was grieved at the thought of losing all his faithful servants for the sake of one man, and he wished heartily24 that he had never set eyes on him, or that he could get rid of him
UDCA is typically used as the initial treatment for PFIC3[29].Some studies have shown that a dose of 10-30 mg/kg/day can successfully treat PFIC3 and resolve the presence of cholestasis in patients[2].UDCA has been shown to be effective in two-thirds of PFIC3 patients.Korkut[30]reported that UDCA treatment was effective in improving conception in women who had intrahepatic cholestasis and were infertile.Here,the patient successfully became pregnant after UDCA treatment following a single IVF treatment,and UDCA treatment may have had some potential beneficial effects.The patient responded well to UDCA therapy.After UDCA treatment,the serum GGT in this patient decreased significantly,and the other liver function indices basically returned to normal.However,after the combination of ICP,UDCA improved the symptoms of pruritus and liver function,but TBA showed an upward trend.The increase in TBA may harm the fetus,so it is necessary to closely monitor the fetal index and deliver early if necessary.The remaining one-third of patients may require additional intervention due to the degree of disease progression and inadequate symptom relief.When other treatments are unsuccessful,liver transplantation has been shown to be effective in PFIC3 patients[31].However,long-term follow-up to monitor the patient’s liver function is necessary.
CONCLUSlON
We were able to definitively diagnose PFIC3 in a 32-year-old female Chinese patient based on her clinical symptoms,pathological examination,and gene detection.Successful gene detection was essential to the diagnosis.This case illustrates the heterogeneity of genetic mutations.These novelheterozygous mutations have a variety of clinical phenotypes that respond well to UDCA therapy.A genetic predisposition to infertility may also be present in this patient,and this requires further research.The discovery of these new mutations has enriched the information on the clinical features of PFIC3 and contributed to a more comprehensive understanding ofdisease.
Oh! if you like fireworks, Princess, said he; and the next night all the will-o -the-wisps in the country came and danced on the plain, which could be seen from the Princess s windows, and as she was looking out, and rather enjoying the sight, up sprang a frightful49 volcano, pouring out smoke and flames which terrified her greatly, to the intense amusement of the Enchanter, who laughed like a pack of wolves quarrelling
ACKNOWLEDGEMENTS
Birbal said, Make you men count, My lord. If you find more crows it means some have come to visit their relatives here. If you find less number of crows it means some have gone to visit their relatives elsewhere .
Now as magicians lose all their power as soon as they are in prison, the King felt himself much embarrassed at being thus at the mercy of those he had so greatly offended
FOOTNOTES
Liu TF reviewed the literature and was responsible for manuscript drafting and organization of illustrations;He JJ and Wang L analyzed and interpreted the pathological findings,immunohistochemical findings,and genetic mutations;Zhang LY was responsible for revision of the manuscript for important intellectual content;all authors issued final approval for the version to be submitted.
The patient’s vital signs were all stable,and physical examination revealed no noteworthy positive signs.
They were six beautiful children; but the youngest3 was the prettiest of them all; her skin was as clear and delicate as a rose-leaf, and her eyes as blue as the deepest sea; but, like all the others, she had no feet, and her body ended in a fish’s tail
Informed written consent was obtained from the patient for publication of this report and any accompanying images.
The authors declare that they have no conflict of interest.
The authors have read the CARE Checklist(2016),and the manuscript was prepared and revised according to the CARE Checklist(2016).
This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial(CC BYNC 4.0)license,which permits others to distribute,remix,adapt,build upon this work non-commercially,and license their derivative works on different terms,provided the original work is properly cited and the use is noncommercial.See:https://creativecommons.org/Licenses/by-nc/4.0/
China
Tian-Fu Liu 0000-0002-6909-5197;Jing-Jing He 0000-0002-1057-6375;Liang Wang 0000-0003-1620-7682;Ling-Yi Zhang 0000-0002-0434-7533.
Ma YJ
Webster JR
You don t know about that, said the youth; I know well enough what it is that lies so heavy on your mind, and I know also of a plan to get the money paid
Ma YJ
 World Journal of Clinical Cases2022年6期
World Journal of Clinical Cases2022年6期
- World Journal of Clinical Cases的其它文章
- Vaginal enterocele after cystectomy:A case report
- Acute kidney injury due to intravenous detergent poisoning:A case report
- Bilateral pneumothorax and pneumomediastinum during colonoscopy in a patient with intestinal Behcet’s disease:A case report
- Successful embolization of an intrahepatic portosystemic shunt using balloon-occluded retrograde transvenous obliteration:A case report
- lmplant site development using titanium plate and platelet-rich fibrin for congenitally missed maxillary lateral incisors:A case report
- Primary duodenal dedifferentiated liposarcoma:A case report and literature review