新型兩性殼聚糖的制備及結構表征
2022-03-24 13:21:02李廣發梁倩瑜梁攸靈莫格格胡章余佳音黃德堅
應用化工 2022年1期
關鍵詞:殼聚糖
李廣發,梁倩瑜,梁攸靈,莫格格,胡章,余佳音,黃德堅
(廣東海洋大學 化學與環境學院 應用化學系,廣東 湛江 524088)
殼聚糖是由來源豐富的甲殼素脫乙酰化得到的一種堿性聚電解質多糖[1-3]。由于具有生物降解性、生物兼容性、安全無毒、廣譜抗菌等優良特性,殼聚糖已廣泛應用于諸多領域[4-6]。但由于殼聚糖僅溶于酸性水溶液,使得其在很多方面的應用受限。由于殼聚糖鏈上含有較多活性基團,通過對殼聚糖上的C6-OH和C2-NH2進行化學改性,可形成不同結構和不同性能的殼聚糖衍生物[7-8]。兩性殼聚糖鏈上具有親水性基團和疏水性基團的特殊結構,在醫藥領域,可增強靶向性給藥,提高藥物的吸收率和生物利用度[9-11]。針對兩性殼聚糖進行更深入的研究,其必將發揮更加重要的作用,成為一種重要的資源。
1 實驗部分
1.1 材料與儀器
甲殼素(分子量50 kDa),生物試劑;2-氯乙胺鹽酸鹽、檸檬醛、氫氧化鈉、尿素、乙酸、乙醇均為分析純。
HJ-A4四位磁力攪拌水浴鍋;FD-1B-50真空冷凍干燥機;GM-0.33A隔膜真空泵;EX125DZH型電子天平;3H20RI智能高速冷凍離心機;P4紫外-可見分光光度計;Spectrum 100傅里葉變換紅外光譜儀;Rigaku Ultima IV組合式多功能X射線衍射儀。
1.2 季銨化甲殼素的制備
取甲殼素粉末1 g,分散在氫氧化鈉溶液中(質量分數為48%,13 g),室溫攪拌24 h;加入適量的冰水,劇烈攪拌直至為均勻溶液,此時溶液中甲殼素和NaOH的質量分數分別為1.3%和8.0%。于80 ℃攪拌下,分批次緩慢加入2-氯乙胺鹽酸鹽5.65 g,磁力攪拌反應12 h后,采用蒸餾水室溫透析3 d,-20 ℃ 預凍過夜,真空冷凍干燥,得到白色粉末固體,產率為83.56%。
1.3 季銨化殼聚糖的制備
取上述制備的季銨化甲殼素1 g,加入氫氧化鈉溶液(質量分數48%,20 g),于65 ℃磁力攪拌 10 h;蒸餾水室溫透析3 d,中間間歇性更換蒸餾水,直至透析液近似呈中性,-20 ℃預凍過夜,真空冷凍干燥,得到白色粉末固體,產率為63.75%。
1.4 兩性殼聚糖的制備
取季銨化殼聚糖1 g溶解在2%乙酸中(v/v,50 mL),室溫攪拌6 h成透明溶液,利用恒壓滴液漏斗逐滴加入檸檬醛的乙醇溶液(1.86 mmol/L,10 mL),室溫繼續攪拌24 h,加入氫氧化鈉溶液(質量分數為5%),離心收集沉淀,先后用無水乙醇和50%乙醇洗滌沉淀,-20 ℃預凍過夜,真空冷凍干燥,得到類白色粉末固體,產率為79.81%。
1.5 結構表征
1.5.1 UV 甲殼素樣品采用10%氫氧化鈉/4%尿素(W/V)的水相溶劑體系溶解;殼聚糖樣品溶于一定量的醋酸溶液(1%,V/V),參比溶液采用各自相對應的溶劑體系,掃描在波長200~500 nm的吸光度。
1.5.2 FTIR 采用溴化鉀壓片法,取1~2 mg樣品在瑪瑙研缽中研磨成細粉末,再與100 mg干燥的溴化鉀粉末混合均勻,裝入模具內,在壓片機上壓制成片。采用純溴化鉀片去除背景干擾,在 4 000~400 cm-1范圍內掃描,測得樣品紅外光譜。
1.5.3 XRD 將樣品放在X射線衍射儀上掃描,電壓為40 kV,電流為40 mA,CuKα譜線,掃描范圍為5~50°,掃描速度為15 s/步。
2 結果與討論
2.1 兩性殼聚糖的合成機理
在對殼聚糖C6-OH進行化學改性時,經典的方法是對殼聚糖C2-NH2進行保護-脫保護。避開傳統的C6-OH殼聚糖衍生物制備途徑,以甲殼素為起始原料,利用甲殼素C2-乙酰氨基的保護狀態,直接化學改性C6-OH,兩性殼聚糖的合成途徑見圖1。
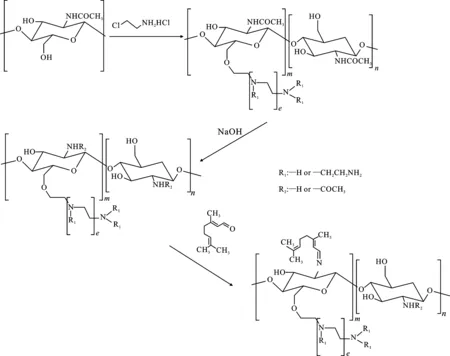
圖1 兩性殼聚糖的合成途徑
氯乙胺鹽酸鹽的毒性較弱,故將其作為接枝多氨基的試劑是很好的選擇。首先,甲殼素的6-OH與ACH通過親核取代反應,得到季銨化的甲殼素;接著,在氫氧化鈉的作用下,進行脫乙酰化反應,得到季銨化殼聚糖;最后與一種羰基試劑(檸檬醛)進行親核加成-消除反應,得到兩性殼聚糖。
2.2 紫外光譜分析

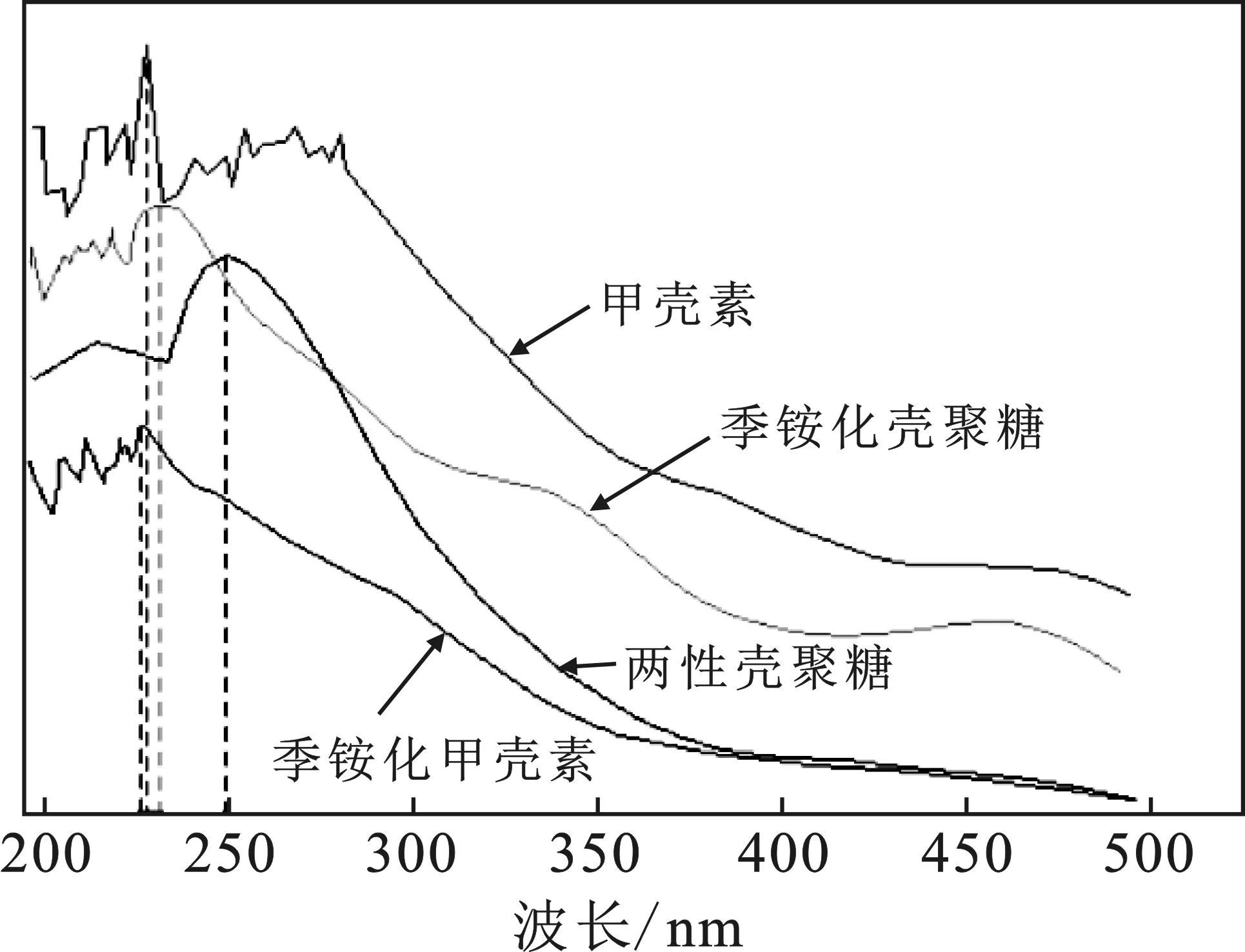
圖2 樣品的紫外光譜圖
2.3 紅外光譜分析
紅外光譜對分子的化學結構敏感,適用于測定不同狀態、濃度和環境下的高分子,是確定高分子聚合物化學結構發生變化的有效工具[12]。由圖3a可知,3 500~3 200 cm-1寬度峰是甲殼素O—H和N—H的伸縮振動疊加峰;2 960~2 830 cm-1屬于飽和C—H的伸縮振動峰;1 150~1 020 cm-1屬于醚鍵C—O的伸縮振動峰。波數1 660,1 558,1 310 cm-1分別對應甲殼素中酰胺Ⅰ、Ⅱ、Ⅲ譜帶的特征吸收峰。波數在3 480,3 264,2 960~2 840 cm-1附近,甲殼素、季銨化甲殼素、季銨化殼聚糖三者的吸收峰強弱有所不同,但峰型基本相似,差異主要在于酰胺的Ⅰ、Ⅱ、Ⅲ譜帶。
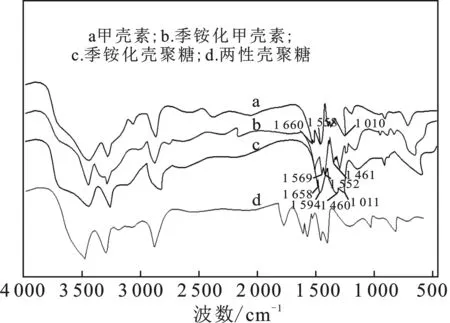
圖3 樣品的紅外光譜圖
季銨化甲殼素與甲殼素的紅外譜圖相比,季銨化甲殼素在1 569 cm-1和1 461 cm-1處出現了兩個新的吸收帶,這與 —NH2基團和 —CH2基團的彎曲振動有關,表明乙氨基團成功接枝到甲殼素分子上。
季銨化殼聚糖與季銨化甲殼素的紅外譜圖相比,季銨化殼聚糖的酰胺Ⅰ和酰胺Ⅲ的吸收帶明顯減弱,酰胺Ⅱ的譜帶消失,并在1 594 cm-1處出現了一個新的吸收譜帶,即 —NH2基團的振動譜帶。綜上表明,季銨化殼聚糖已成功合成。
2.4 X-射線衍射分析
研究物質結構的分析方法很多,但應用最廣泛和最有效的手段是X射線衍射技術,特別是在高分子化學、材料化學等領域中,已成為一種重要的結構分析手段[13]。
由圖4可知,甲殼素在9°和19°處各有一個明顯的特征衍射峰,經甲殼素逐步合成反應后,甲殼素的9°和19°特征衍射峰發生了明顯的變化,其中每步反應產物的9°特征衍射峰均在減弱,到最后形成兩性殼聚糖時消失。而位于19°處的特征衍射峰從第二步產物季銨化殼聚糖開始明顯變弱,成為較寬的峰。甲殼素、季銨化甲殼素和季銨化殼聚糖三種物質的衍射峰出峰情況較為類似。此外,甲殼素和第一步產物季銨化甲殼素在26°都出現小的峰形,經第二步反應后該峰形變得模糊,到第三步反應產物后,該小峰就消失了。這表明殼聚糖鏈在一些方向具有一定的規整性,但是結晶度低,而經過化學改性之后的殼聚糖,在形成氫鍵的能力方面大大下降,一部分結晶型轉變成了無定型。
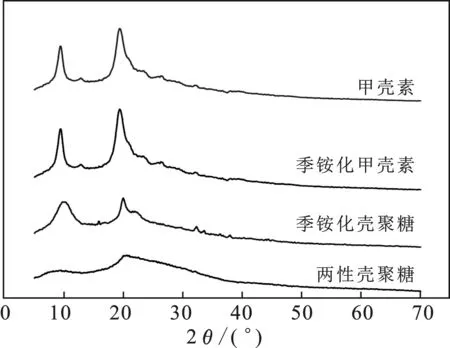
圖4 樣品的X射線衍射圖
3 結論
本文以甲殼素為原料,通過C6-OH與氯乙胺鹽酸鹽發生親核取代反應,接著對C2-氨基脫乙酰化,隨后通過氨基與羰基發生希夫堿反應,制備得到兩性殼聚糖。通過紫外、紅外、X-射線衍射分析,一種新型兩性殼聚糖成功合成。該制備方法回避了C6-OH殼聚糖的常規衍生途徑,即C2-氨基的保護-脫保護,制備工藝更加簡便。
猜你喜歡
河北科技師范學院學報(2022年2期)2022-08-26 08:55:40
河北科技師范學院學報(2021年1期)2021-05-10 03:34:20
中成藥(2017年12期)2018-01-19 02:06:57
電源技術(2017年1期)2017-03-20 13:37:59
廣西科技大學學報(2016年1期)2016-06-22 13:10:38
天然產物研究與開發(2016年1期)2016-06-05 10:29:25
食品界(2016年4期)2016-02-27 07:36:46
中國果菜(2015年2期)2015-03-11 20:01:01
應用化工(2014年7期)2014-08-09 09:20:21
應用技術學報(2014年4期)2014-02-28 14:52:40
