低氧致肺動脈平滑肌細胞鈣穩態失衡及藥物干預研究進展
2022-04-07 02:48:04蓋祥云趙恩麒王金宇趙悅孚何彥峰林鵬程
中國藥理學通報 2022年4期
關鍵詞:研究
蓋祥云,趙恩麒,王金宇,趙悅孚,何彥峰,林鵬程
(青海民族大學1. 藥學院、2. 青海省青藏高原植物化學重點實驗室,青海 西寧 810007)
慢性缺氧性肺疾病(支氣管擴張、慢性阻塞性肺疾病、特發性肺纖維化和限制性胸壁異常等),是世界上最常見的致殘和死亡原因之一,低氧性肺動脈高壓(hypoxic pulmonary hypertension,HPH)是導致肺部血管阻力升高的主要機制[1]。HPH的發生發展經歷了急性期的低氧性肺血管收縮(hypoxic pulmonary vasoconstriction,HPV)以及慢性期的低氧性肺血管重塑(hypoxic pulmonary vascular remodeling,HPVR)[2]。HPH通常早期難以發現,直到后期出現心力衰竭,如呼吸困難、心悸和下肢水腫等癥狀才會被診斷治療[3]。該疾病的發病機制復雜,目前的研究尚未能完全解釋其發生發展機制。以往研究發現:肺動脈平滑肌細胞(pulmonary artery smooth muscle cells,PASMCs)內游離鈣離子濃度(cytoplasmic free Ca2+concentration,[Ca2+]i)異常升高是肺血管持續收縮和重塑導致肺動脈高壓發生的主要誘因[4-5]。低氧條件下,諸多調節途徑參與誘導細胞內Ca2+水平的升高,繼而引發細胞鈣穩態失衡。一方面,作為PASMCs內最重要和最根本的第二信使,Ca2+不僅參與了HPV的形成和發展過程,也能夠通過調節PASMCs的遷徙與增殖,促進HPVR的形成[6-7];另一方面,鈣信號轉導通路在維持肺動脈內皮細胞屏障完整性以及肺動脈內皮細胞釋放舒血管物質等過程中都發揮了重要作用[3]。
本文著眼于低氧導致PASMCs鈣穩態失衡的作用機制,就近年來的相關研究做一綜述。全文共分為3個主要部分:低氧與PASMCs鈣穩態失衡;鈣穩態失衡引發HPV的機制;鈣穩態失衡在HPVR中的作用。
1 低氧與PASMCs鈣穩態失衡
1.1 低氧-缺氧誘導因子-1(hypoxia-inducible factor-1, HIF-1)-PASMCs鈣穩態失衡低氧可誘導PASMCs中HIF-1的聚集。HIF-1由α和β亞基構成。HIF-lα是氧調節蛋白,為HIF-1的特有組份,而HIF-1β是多種轉錄因子的共同亞基。常氧下,胞質中HIF-1α易降解。相反,缺氧時HIF-1α的降解受到抑制。HIF-1α在胞質中聚積,并與HIF-1β形成穩定的異二聚體。HIF-1聚積,使編碼鈣庫操縱性鈣通道和受體操縱性鈣通道的經典瞬時受體電位通道1(transient receptor potential canonical 1,TRPC1)和經典瞬時受體電位通道6(transient receptor potential canonical 6,TRPC6)表達水平上調,誘導鈣內流,胞內游離鈣水平升高,最終促進HPV和HPVR的發生[8]。前期研究發現,在低氧條件下,敲除HIF-1α的大鼠,發生肺動脈高壓的幾率顯著下降,肺動脈重構明顯改善[9]。近年研究發現,低氧條件下,敲除HIF-1α能夠下調PASMCs中的TRPC1和TRPC6通道的表達和功能[10]。慢性低氧條件下,白楊素能夠通過下調PASMCs中HIF-1α、TRPC1和TRPC6的表達來調節細胞內鈣濃度,從而發揮改善肺動脈高壓的作用[11]。
1.2 低氧-K+通道-PASMCs鈣穩態失衡低氧條件下,PASMCs上的電壓依賴性K+通道失活,K+外流受限制,細胞膜發生去極化,進而使得電壓依賴性鈣通道開放,[Ca2+]i增加。Ca2+通過與肌質網質膜上的鈣通道蛋白蘭尼堿受體(ryanodine receptors,RyR)結合,進一步誘導肌質網內鈣釋放,觸發細胞興奮收縮耦聯機制,導致平滑肌收縮,從而促進HPV[6]。Hua等[12]研究發現,蘋果多酚能夠通過影響K+通道活性,減少PASMCs胞質內的游離鈣濃度,從而逆轉肺血管收縮[12]。
1.3 低氧-鈣庫操縱性鈣通道(store-operated calcium channel, SOC)-PASMCs鈣穩態失衡SOC可調控內質網(endoplasmic reticulum,ER)/肌漿網(sarcoplasmic reticulum,SR)鈣庫耗竭而引發的[Ca2+]i升高,稱為鈣庫操縱性鈣內流。另外,鈣庫耗竭,SOC可誘導鈣庫的重新充盈。目前的研究發現在PASMCs中,調控Ca2+參與低氧反應的主要是SOC中的TRPC1和TRPC6,另外也有研究表明Orai 1蛋白也參與其中[13]。
基質交感分子1 (stromal interaction molecule,STIM1)主要參與調控鈣庫操縱性鈣內流。在正常狀態下,STIM1作為一種單通道跨膜蛋白定位于ER/SR膜,當ER/SR的內鈣耗竭時,STIM1離開ER/SR膜,轉運至細胞膜,與Orai 1通道形成鈣釋放激活鈣通道(Ca2+release-activated Ca2+current,CRAC),它對鈣高度敏感,能夠促進外鈣內流。隨著細胞內鈣庫再次充盈,STIM1失活,鈣內流減少[14]。
在眾多的通路中,低氧通過SOC誘導鈣庫操縱性鈣內流增加是PASMCs的[Ca2+]i升高的重要機制之一。研究表明:低氧處理,能夠使得PASMCs表面的SOC表達增加,繼而通過SOC的外鈣內流增加[15]。另外,低氧條件下,人肺動脈內皮細胞中經典瞬時受體電位通道4轉錄水平升高,使得SOC表達增加、功能上調,細胞內鈣水平升高,進一步使激活蛋白1活性及表達均增加。同時,血管內皮生長因子、內皮素1(endothelin-1,ET-1)及血小板衍生生長因子等應答基因的表達水平升高,最終誘導肺動脈內皮細胞的生長因子增加,從而促進PASMCs的增殖,導致肺動脈重構[16]。Wang等研究發現西地那非能夠通過下調TRPC1的表達,抑制PASMCs的鈣庫操縱性鈣內流,從而改善缺氧誘導的PASMCs增殖[17]。
1.4 低氧-受體操縱性鈣通道(receptor-operated calcium channel,ROC)-PASMCs鈣穩態失衡目前的研究表明:ROC定位于細胞膜上,在PASMCs中,ROC由經典瞬時受體電位通道3、TRPC6、經典瞬時受體電位通道7參與構成。低氧條件下,ET-1、5-羥色胺(5-hydroxytryptamine,5-HT)、血管緊張素Ⅱ、血小板生長因子等血管活性物質與G蛋白偶聯受體結合,激活磷脂酶C(phospholipase,PLC),PLC能夠進一步催化二磷酸磷脂酰肌醇裂解成三磷酸肌醇(inositol 1, 4, 5-triphosphate,IP3)、二酰基甘油(diacylglycerol,DAG)。IP3與SR或ER膜上的IP3受體(IP3 receptor,IP3R)結合,導致Ca2+從SR/ER釋放或動員進入細胞質,[Ca2+]i升高;DAG能夠誘發ROC的開放,DAG作為第二信使,能夠直接激活ROC,[Ca2+]i升高[14]。研究表明:U73122作為PLC抑制劑,能夠減少DAG的生物合成,從而減弱野生型小鼠HPV;另外,使用OAG(DAG類似物)激活TRPC6或者減少DAG的降解,都能夠引發小鼠發生肺動脈收縮[18]。Kaymak等[19]研究表明,在低氧誘導的實驗性肺動脈高壓大鼠中,氯喹能夠降低肺動脈中TRPC1、TRPC6和鈣敏感受體的表達,從而改善肺動脈重構。
1.5 低氧-鈣敏感受體(Ca2+-sensing receptor,GaSR)-PASMCs鈣穩態失衡Ca2+作為第二信使在HPV和HPVR中發揮重要作用的同時,又是其中關鍵的第一信使。低氧處理后,細胞外鈣能夠通過激活細胞膜上的GaSR,進一步激活PLC-IP3-Ca2+途徑,IP3與PASMCs內鈣庫上的IP3受體結合,進一步引發內鈣釋放,胞內的第二信使Ca2+水平升高。
CaSR是一種G蛋白偶聯受體,能夠感知細胞外鈣濃度的變化,參與到鈣穩態調節過程中[20]。目前的研究發現:CaSR在血管平滑肌細胞和內皮細胞中均有表達[21],它能夠被氨基酸(苯丙氨酸和谷氨酸)、多價陽離子(Ca2+,Gd3+,Mg2+)以及氨基糖苷類抗生素等激活。Yamamura等[22]的研究表明:在低氧導致的肺動脈高壓小鼠和野百合堿引發的肺動脈高壓大鼠中,PASMCs的CaSR表達水平均升高;利用NPS2143阻斷鈣敏感受體后,HPH動物的右心室收縮壓、右心肥大指數均顯著降低,并且肺小動脈的重構得到明顯改善。
1.6 低氧-低氧誘導促有絲分裂因子(hypoxia-induced mitogenic facto,HIMF)-PASMCs鈣穩態失衡HIMF具有促有絲分裂、血管生成、血管收縮和趨化因子樣的特性,這些特性都與病理性肺疾病(血管收縮、血管重塑、炎癥和纖維化)有關。2009年,Fan等[23]發現HIMF能夠通過激活PLC-IP3途徑,引發人PASMCs內鈣水平升高,導致肺血管收縮,最終引發肺動脈高壓。HIMF為PASMCs外CaSR的非經典配體,低氧刺激下HIMF與CaSR結合能夠引發PASMCs增殖和HPH的發生[24]。最新的研究證明,在HIMF誘導肺動脈高壓形成中,高遷移率族蛋白B1-晚期糖基化終末產物受體信號轉導通路在“肺動脈平滑肌細胞-內皮細胞”相互調控的過程中發揮著關鍵作用[25]。
低氧導致PASMCs的[Ca2+]i升高的可能機制(Fig 1)有待進一步的研究和完善,隨著今后研究的深入,會發現更多的調控通路和潛在的藥物靶點。
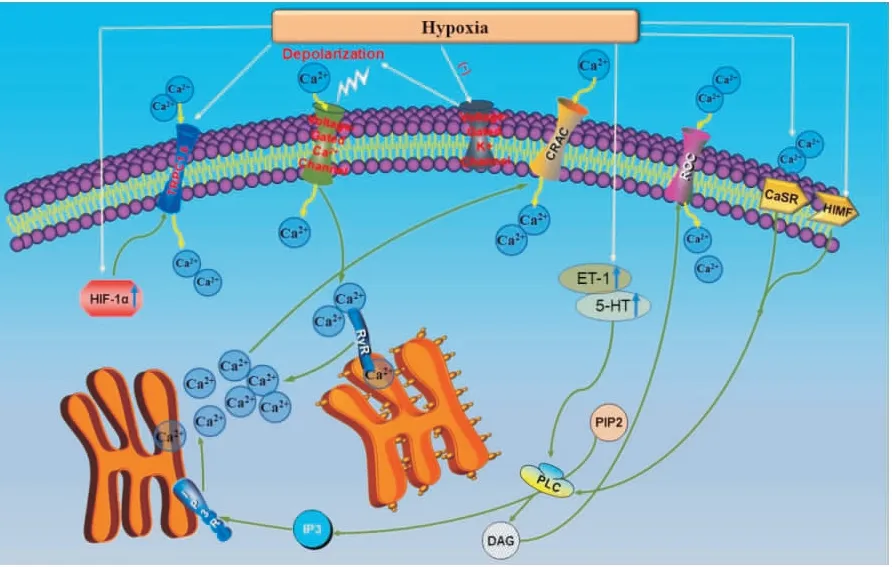
Fig 1 Mechanism of hypoxic-induced increase of intracellular free calcium concentration in PASMCs
2 HPV與PASMCs鈣穩態失衡
HPV是指低氧刺激下,眾多因素導致的強烈的肺動脈收縮。低氧下,HPV這一特殊的反應是機體為了維持肺組織正常的通氣/血流比值所產生的適應機制,但是隨著低氧的持續發生,這一反應也會變成引發肺動脈高壓的病理因素[1]。低氧條件下,HPV發生發展的一項重要誘因就是“低氧導致的PASMCs內鈣水平升高”[26]。
如前所述,低氧能夠通過諸多途徑引發PASMCs內游離鈣水平升高,細胞內鈣離子進一步與鈣調蛋白(calmodulin,CaM)結合形成Ca2+/CaM復合物,激活肌球蛋白輕鏈磷酸酯酶,引發肌球蛋白輕鏈磷酸化,進一步激活肌動蛋白-肌球蛋白收縮裝置,導致PASMCs發生收縮,引發HPV。
3 HPVR與PASMCs鈣穩態失衡
HPVR是指PASMCs增殖、細胞外基質增加等導致的動脈管壁增厚及肺小動脈管腔狹窄。目前,針對HPH中HPVR的治療方法有限,肺血管重塑的重要驅動因素之一就是PASMCs的過度增殖和凋亡抵抗[27]。降低PASMCs內[Ca2+]i可以有效地抑制或者改善HPVR,使得肺動脈壓力降低[7]。
3.1 [Ca2+]i升高-PI3K/Akt/mTOR-PASMCs增殖Akt又被稱為蛋白激酶B,是磷脂酰肌醇-3激酶(phosphatidylinositol 3-kinase,PI3K)下游的作用位點之一。作為與PI3K/Akt通路密切關聯的蛋白激酶之一,雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)可作為Akt下游的一個底物被激活。
PI3K/Akt/mTOR可參與細胞凋亡、細胞增殖、細胞自噬等多個環節。Lee等[28]證實,低氧可以激活雞肝細胞Ca2+/PI3K/Akt/mTOR通路,調節細胞周期蛋白,促進雞肝細胞DNA合成,從而誘導細胞增殖。PASMCs內游離鈣水平升高,可激活PI3K/Akt/mTOR,使得細胞周期蛋白表達增加,最終誘導PASMCs的增殖[23]。研究發現,慢性低氧誘導的肺動脈高壓小鼠模型中,低氧誘導HIMF表達增加,誘導Akt磷酸化,能夠促進PASMCs增殖[29]。
3.2 [Ca2+]i升高-PKC/MAPKs-PASMCs增殖蛋白激酶C(protein kinase C,PKC)由Ca2+激活,調節細胞凋亡及生長分化等過程。絲裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK),包括p44/42MAPK、p38MAPK(α、β)、Jun N端激酶等,可被多種細胞外信號激活。MAPK被活化,轉移至細胞核內,能夠調節細胞周期蛋白。缺氧能夠通過誘導雞肝細胞[Ca2+]i升高,激活PKC/MAPKs,進一步促進DNA合成,誘導細胞發生異常增殖[28]。目前的研究表明:缺氧通過誘導大鼠PASMCs的HIMF表達水平上調,進一步引發細胞內Ca2+水平升高,通過調控PASMCs中PKC/MAPKs信號轉導通路,從而誘導細胞異常增殖[30]。
4 總結與展望
綜上所述,低氧導致PASMCs鈣穩態失衡過程中,HIF-1α、TRPC1、TRPC6、部分敏感的K+通道、PLC、CaSR、HIMF及ROC相關的膜受體都有可能成為HPH防治藥物的潛在靶點或者干預目標。
目前為止,雖然越來越多的研究者關注Ca2+在HPH發生發展過程中扮演的角色,但是仍然存在著很多不明確的問題。首先,低氧誘導PASMCs發生鈣穩態失衡的相關機制并不是單獨存在的幾條通路,而是一個復雜的網絡機制,目前的研究并不能完全、清楚地闡述這個網絡機制;第二,鈣超載引發HPV和HPVR還涉及哪些因素和信號通路,也有待研究;第三,低氧后,PASMCs鈣超載與肺動脈內皮細胞的損傷有哪些聯系,也需要進一步研究和解釋。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
