Theoretical study on the improvement of the doping efficiency of Al in 4H-SiC by co-doping group-IVB elements
2022-04-12 03:47:08YuanchaoHuang黃淵超RongWang王蓉YixiaoQian錢怡瀟YiqiangZhang張懿強(qiáng)DerenYang楊德仁andXiaodongPi皮孝東
Chinese Physics B 2022年4期
Yuanchao Huang(黃淵超) Rong Wang(王蓉) Yixiao Qian(錢怡瀟) Yiqiang Zhang(張懿強(qiáng))Deren Yang(楊德仁) and Xiaodong Pi(皮孝東)
1State Key Laboratory of Silicon Materials and School of Materials Science and Engineering,Zhejiang University,Hangzhou 310027,China
2Hangzhou Global Scientific and Technological Innovation Center,Zhejiang University,Hangzhou 311200,China
3School of Materials Science and Engineering&Henan Institute of Advanced Technology,Zhengzhou University,Zhengzhou 450001,China
Keywords: 4H-SiC,p-type,co-doping,ab initio study
1. Introduction
As a wide bandgap semiconductor, silicon carbide(SiC)has been known as a good material for semiconductor devices since early the 20thcentury.[1-3].Among all kinds of SiC polymorphs,4H-SiC has attracted great attention given its superior electrical properties.[1]The effective n-type doping of 4H-SiC may be readily carried out by doping nitrogen (N) because the ionization energy of N is rather small (~0.06 eV).[4-6]However,the efficiency of the p-type doping of 4H-SiC has remained poor despite its long-time development.This is mainly attributed to the large ionization energies of p-type dopants in 4H-SiC.For example,the most widely used p-type dopant of aluminum(Al)has the ionization energy of~0.23 eV,which leads to the ionization rate of no more than~30%for the typical doping concentration of Al in the range from 1016cm-3to 1018cm-3at room temperature.[7-11]The low ionization rate of Al means that a large amount of unionized Al is in 4H-SiC,limiting the mobility of holes by impurity scattering.[9]In addition, the carrier capture/emission of unionized Al poses severe reliability issues for devices based on 4H-SiC.[8]Hence,the poor doping efficiency of p-type dopants in 4H-SiC needs to be well addressed, especially given the fact that the development of 4H-SiC has recently gained great momentum for high power electronics.[1-3]A series of important 4H-SiC devices such as p-type SiC metal-oxide-semiconductor fieldeffect transistors(MOSFETs)[12]and n-channel insulated gate bipolar transistors(IGBTs)[13-15]demand that 4H-SiC should be much more effectively doped with p-type dopants.
Various approaches such as host alloying,[16-19]strain modulation,[20]incorporation of band-like defect-induced energy levels[21]and co-doping[22-30]have been proposed to lower the ionization energies of dopants in wide-bandgap semiconductors. Among these approaches, co-doping is attractive due to its simple process and negligible damage to the host. When a p- or n-type dopant is co-doped with other dopants, defect-level repulsion may push the energy level of the p-or n-type dopant to a shallower position,decreasing the ionization energy of the p- or n-type dopant. The effectiveness of co-doping strongly depends on the symmetries of the energy levels of dopants. By taking advantage of co-doping,researchers have successfully improved the efficiency of the n-type doping of diamond[22-24]and ZnTe[25]and the p-type doping of ZnO.[26-30]As for p-type doping of 4H-SiC, the acceptor-donor co-doping,forming a A2D complex,has been studied usingab initiocalculations.[31]It shows that acceptordonor co-doping can improve the solid solubility of acceptor,but it cannot lower down the ionization energies effectively,because the energy levels of the acceptor and donor have different atomic characteristics and symmetries. An effective way to lower the ionization energies p-type doping of SiC has been not obtained so far.
In this study, we demonstrate that acceptor and group-IVB elements co-doping is an effective means to improve the doping efficiency of the most widely used p-type dopant,i.e.,Al,in 4H-SiC.By analyzing the symmetry of the energy levels of elements throughout the whole periodic table in the framework of crystal field theory, we first identify that the energy levels of group-IVB elements have the same symmetry as that of Al. The coupling between the energy levels of group-IVB elements and that of Al should push the energy level of Al towards the valance band maximum(VBM),reducing the ionization energy of Al. Subsequent first-principles calculations verify that the unoccupied energy levels of group-IVB elements indeed interact with the acceptor level of Al,effectively lowering the ionization energy of Al. We find that Ti has the most prominent effectiveness among all the group-IVB elements owing to its lowest orbital energies and smallest atomic size. The co-doping of Ti decreases the ionization energy of Al by nearly 50%, leading to a value as low as~0.13 eV.As a result, the ionization rate of Al with Ti co-doping may be up to~5 times larger than that without co-doping at room temperature when the concentration of Al is up to 1018cm-3.
2. Symmetry analysis
It is well known that Al prefers substituting Si in 4HSiC.[11]As shown in Fig. 1(a), the orbital of Al splits into a fully occupied single a1state and a 3/4 occupied doubledegenerated e state in theC3vsymmetry of 4H-SiC.The e state can capture an electron from the VBM of 4H-SiC,enabling the p-type doping of Al. In an ideal co-doping case,an impurityXshould introduce an upper unoccupied e state in the bandgap of 4H-SiC. The coupling between the e states ofXand Al pushes the e state of Al to a shallower position,improving the efficiency of the p-type doping of Al. The symmetry of substitutionalXcan be predicted by the crystal field theory.[32]Furthermore, by comparing the number of valence electrons ofX, we classifyXin the periodic table into three groups,which are referred to as group-A elements(with less than four valence electrons),group-B elements(with more than four valence electrons)and group-C elements(with four valence electrons).
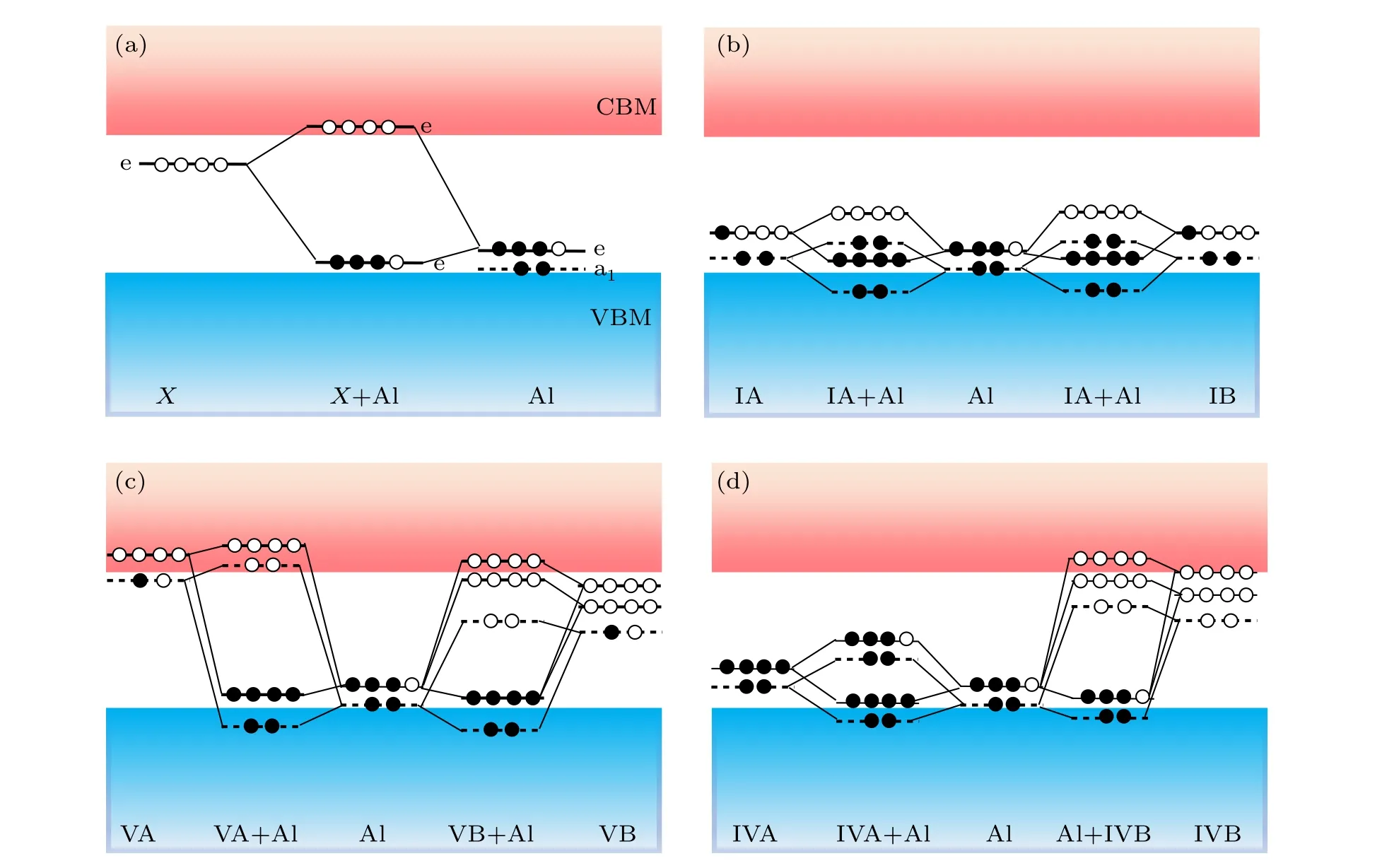
Fig.1. (a)Schematic illustration of the defect-level repulsion between the defect states of AlSi and an ideally substitutional impurity X with an upper empty e state. The analysis of defect-level repulsion between the defect states of AlSi and substitutional group-A impurities(b),group B impurities(c),as well as group-C impurities(d)are carried out based on the crystal field theory.
Group-A elements include group-IA, IIA, IIIA elements and group-IB, IIB, IIIB elements. The orbital energies of group-A elements are higher than that of Si (or C), and the number of valence electrons is less than four. When group-A elements substitute host atoms,the defect energy levels are created from the host valence band states that move upward in energy.[33]Taking group-IA with one valance electron as an example, the defect levels of substitutional group-IA impurities consist of a low-lying fully occupied singlet a1state and a 1/4 occupied double-degenerated e state near the VBM[Fig. 1(b)]. After co-doping with Al, defect-level repulsion may occur between the 1/4 occupied e state of substitutional group-IA impurities and the 3/4 occupied e state of AlSi. The electron on the e state of substitutional group-IA impurities will transfer to the 3/4 e state of Al,which gives rise to a lower fully occupied e state and an unoccupied e state [Fig. 1(b)].This increases the ionization energy of the acceptor. The symmetry of the defect level is the same for the other elements belongs to group-A elements,but more electrons would occupy the highest e state. Therefore, group-A elements may not be used as co-dopants for Al in 4H-SiC.
Group-B elements contain group-VA, VIA, VIIA and group-VB, VIB, VIIB, VIIIB elements. Because the orbital energies of group-B elements are higher than that of Si (or C),the defect energy levels are created from the host conduction band states that move downward in energy.[33]For substitutional group-VA impurities, the a1state and e state move downward near the CBM, and one electron occupies the a1state[Fig.1(c)]. After co-doping with Al,the electron on the a1state of substitutional group-VA will transfer to the 3/4 occupied e state of AlSi.Substitutional group-VA impurities have more than one electron in the defect levels, one of the electrons transfers to the e state of AlSiand the other will occupy the higher a1state [Fig. 1(c)]. This leads to the formation of donor states when VIA(or VIIA)elements are co-doping with Al, which indicates that VIA (or VIIA) elements are not for the choice of co-doping. For group-VB,VIB,VIIB,VIIIB elements, the d orbital splits into one a1state and two e states in theC3vsymmetry of 4H-SiC,so the defect level has one a1state and two e states. After co-doping with Al, the electron in the defect state of substitutional group-VB impurities passivates the e state AlSi. Similarly, co-doping of group-VIB,VIIB and VIIIB elements with Al acts as a donor. Therefore,group-B elements are not ideal co-dopants of Al.
Group-C elements contain group-IVA and group-IVB elements with the same number valence electrons as SiC.Since the p orbital energies of Ge, Sn, and Pb are higher than that of Si. When substituting Si, the defect states are created from the host valence band states that move upward in energy, which contain fully occupied a1state and e state near VBM[Fig.1(d)]. This occupied e state would repel the e state of AlSitoward a higher energy position in the bandgap,which increases the ionization energy of AlSi.For group-IVB,the valence electrons of group-IVB elements have the d2s2arrangement. In theC3vsymmetry of 4H-SiC,the d orbital splits into one full occupied a1state and two unoccupied e states. Because the d orbital energy is lower than that of the p orbital of Si,[34]the defect states of substitutional group-IVB impurities are created from the host conduction band states that move downward in energy, which contain one unoccupied a1state and two unoccupied e states. Therefore, the group-IVB elements are of great potential to lower the e state of Al by taking advantage of defect-level repulsion between the e states of substitutional group-IVB impurities and AlSi, and decreasing the ionization energy of Al in 4H-SiC.
3. First-principles calculations
Now we carry out first-principles calculations to examine the effect of the co-doping of group-IVB elements such as Ti,Zr and Hf on the ionization energy of Al in 4H-SiC.The firstprinciples calculations are performed by using the Viennaabinitiosimulation package (VASP). The generalized gradient approximation(GGA)in with the Perdew,Burke,and Ernzerhof (PBE) functionals is employed to describe the exchangecorrelation interactions.[35]A 4×4×1 supercell of 4H-SiC with 128 atoms is constructed. A single-doping model is constructed by replacing one Si atom with one dopant atom,while a co-doping model is built by replacing two neighboring Si atoms with two different dopant atoms. The plane-wave energy cut-off is 500 eV. Thek-mesh is set to be 2×2×2.During the structural relaxations a conjugate-gradient algorithm is used. In a relaxed structure the force on each atom is lower than 0.01 eV/°A. The total energy is converged to 1.0×10-6eV. To accurately calculate the bandgap, we use static calculations with the hybrid functional of Heyd,Scusseria and Ernzherof (HSE06) to work on the relaxed structures obtained with the PBE functionals. Our calculation shows that the calculated lattice parameters of 4H-SiC area=3.07 °A andc=10.05 °A. The calculated bandgap of 4H-SiC is 3.18 eV.These are well consistent with experimental results.[1]
The formation energy of a dopant atom at the charge state ofqin 4H-SiC is defined as[36,37]

where ΔE(dopant,q)=E(dopant,q)-E(host)+∑niE(i)+qεVBM.E(dopant,q) is the energy of a supercell containing the dopant atom at the charge state ofq.E(host)is the energy of the host in the same supercell without the dopant.εVBMis the energy of the VBM of the host.EFis the Fermi energy referenced to the VBM.μiis the chemical potential of constituentireferenced to elemental solid or gas with energyE(i).niis the number of atoms removed from or added into the supercell. In this work, two chemical-potential conditions (C-rich and Si-rich)are considered due to the limitation of the formation energy of bulk 4H-SiC.The charge transition energy level with respect to the VBM is calculated in the mixedk-point scheme by using[37]


4. Results and discussion
The calculated total energies suggest negligible difference(within 0.02 eV)between the substitutionalkandhsites of Si for Al, Ti, Zr and Hf. Therefore, we only consider that Al,Ti,Zr,and Hf substitutek-site Si in this work. We first examine the single-electron levels of all the dopants,which actually correspond to the vertical activation energies in Eq. (2). The results are shown in Fig.2. We can see that the fully occupied a1state of Al is located atEV+0.11 eV. The 3/4 occupied e state of Al is located atEV+0.21 eV.For Ti,Zr,and Hf they each introduce an unoccupied a1state and two e states near the CBM,which is consistent with the aforementioned symmetry analysis. WhenXchanges from Ti to Hf, the unoccupied a1state and two e states ofXshift upward because the d orbital energy increases. Among group-IVB elements the states of Ti are the lowest due to its lowest d orbital energy and smallest atomic size. After co-doping Al with Ti, Zr, and Hf we observe that the 3/4 occupied e state of Al shifts toEV+0.11 eV,EV+0.13 eV,andEV+0.15 eV,respectively. The unoccupied a1state and two e states of Ti/Zr/Hf near the CBM are pushed into the conduction band of 4H-SiC.These confirm the effective coupling between Al and group-IVB elements.
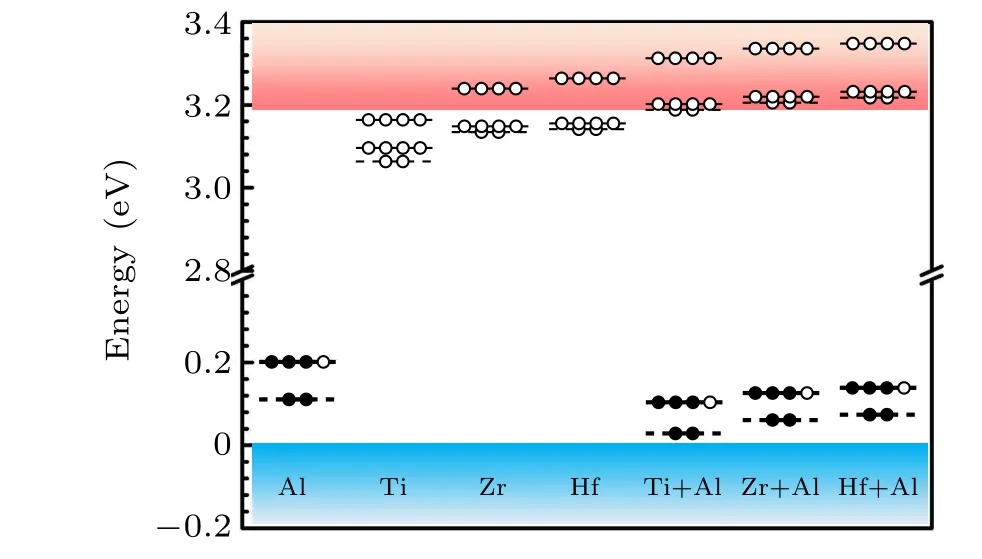
Fig.2. Single-electron levels of Al,Ti,Zr,Hf,Ti+Al,Zr+Al,and Hf+Al in 4H-SiC.All the dopants substitute Si in 4H-SiC.
We have then calculated the charge transition energies and formation energies of Al, Ti, Zr, Hf, Ti+Al, Zr+Al,and Hf+Al, as shown in Fig. 3. The calculated charge transition energies and formation energies of neutral dopants are tabulated in Table 1. The formation energy of neutral Al is 1.9 eV and 2.2 eV at the C-rich limit and Si-rich limit,respectively. The calculated(0/-)transition energy of Al,namely,the ionization energy,isEV+0.23 eV,which is in agreement with experiment results.[1]For Ti, Zr, and Hf the formation energies are 1.08/1.40 eV, 2.27/2.59 eV, and 1.70/2.03 eV at the C-rich/Si-rich limit, respectively. Their (0/-) transition energies areEC-0.17 eV,EC-0.08 eV, andEC-0.06 eV,respectively. Please note that the current results for Ti are very consistent with experimental observation. It was found that Ti in the graphite parts of a 4H-SiC single-crystal growth equipment might be unintentionally incorporated into 4H-SiC,[38,39]indicating the small formation energy of Ti. Daliboret al.once observed a very deep acceptor level atEC-0.17 eV in 4H-SiC by using deep-level transient spectroscopy,[37]exactly agreeing with the currently obtained(0/-)transition energy atEC-0.17 eV for Ti. With the co-doping of Ti, Zr and Hf the ionization energy of Al significantly decreases toEV+0.13 eV,EV+0.16 eV, andEV+0.18 eV, respectively.It is clear that the co-doping of Ti has the most prominent effectiveness, which decreases the ionization energy of Al by nearly 50%. This is due to the lowest d orbital energy of Ti,which induces the strongest defect-level repulsion with Al. In the meantime, the formation of Ti is smaller than that of Al,which can guarantee a sufficient amount of Ti doping in SiC.
The stability of Ti+Al, Zr+Al, and Hf+Al is subsequently checked by calculating their binding energies withEb=ΔHf(IVB)+ΔHf(Al)-ΔHf(IVB+Al). The results are shown in Table 1. The values ofEbfor Ti/Zr/Hf+Al are all positive, indicating that the formation of Ti/Zr/Hf+Al is favorable in energy when Ti/Zr/Hf and Al coexist. Ti/Zr/Hf can form stable complexes with Al, effectively doping 4HSiC.Besides,if the amount of Ti/Zr/Hf doping is greater than Al, it would form isolated Ti/Zr/Hf and inducing an accepter level (0/-). This could not significantly impact the carrier density of Al-doped 4H-SiC, because the isolated Ti is also an accepter. It also cannot significantly impact the carrier lifetime,because the(0/-)level of Ti is close to the CBM,which makes it hard to trap the holes.

Fig.3. Formation energies of Al,Ti,Zr,Hf,Ti+Al,Zr+Al,and Hf+Al at the(a)C-rich limit and(b)Si-rich limit.

Table 1. Charge transition energies, formation energies of neutral defects and binging energies.

Fig. 4. The ionization rate of Al with the co-doping of (a) Ti, (b) Zr,and (c) Hf at different doping concentrations as a function of temperature.The dashed lines and solid lines represent the ionization rate of Al and Ti/Zr/Hf+Al,respectively.

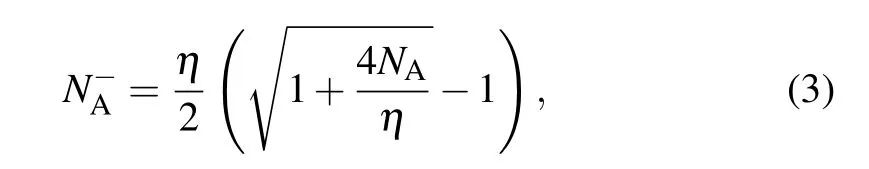
whereηis given byη=NV/gAexp(-ε(0/-)/kT),ε(0/-)is the ionization energy,gAis the degeneracy factor of Al,andNVis the effective density of states in the valence band given byNV= 2(2πm?dhkT/h2)3/2. Please note thatm?dhis the density-of-states effective mass for holes,his the Planck’s constant,kis the Boltzmann constant, andTis the temperature. As shown in Fig. 4, the ionization rate is found to decrease with the increase of the concentration of Al in the typical range from 1014cm-3to 1018cm-3. When only Al is doped, the ionization rate of Al at room temperature is~88%, 56%, 24%, 8%, and 3% for the concentrations of 1014cm-3,1015cm-3,1016cm-3,1017cm-3,and 1018cm-3,respectively. With the co-doping of Ti/Zr/Hf the ionization rate of Al is significantly improved. Among these co-dopants the improvement induced by Ti is the most pronounced. For the Al concentrations of 1014cm-3, 1015cm-3, 1016cm-3,1017cm-3,and 1018cm-3the co-doping of Ti makes the ionization rate of Al reach 100%, 96%, 80%, 44%, and 16% at room temperature, respectively. Clearly, for the highest concentration of 1018cm-3the Ti-induced improvement of the ionization rate of Al is the most significant(a factor of~5).
5. Conclusion
In summary,we have found that group-IVB elements may be employed to co-dope Al, effectively lowering the ionization energy of Al via the coupling between the energy levels of group-IVB elements and that of Al. Among the group-IVB elements Ti most significantly reduces the ionization energy of Al (~50%), leading to a value of~0.13 eV. Such a low ionization energy enables the ionization rate of Al to be up to~5 times larger than that without co-doping at room temperature when the concentration of Al is up to 1018cm-3. Given the fact that group-IVB elements such as Ti have already been incorporated into 4H-SiC during its growth,[38,39]the current work should inspire the intentional doping of group-IVB elements together with Al during the materials preparation of 4H-SiC to effectively improve the doping efficiency of Al.
Acknowledgments
Project supported by the National Key Research and Development Program of China (Grant Nos. 2017YFA0205704 and 2018YFB2200101), the National Natural Science Foundation of China (Grant Nos. 91964107 and 61774133), Fundamental Research Funds for the Central Universities, China(Grant No. 2018XZZX003-02), the National Natural Science Foundation of China for Innovative Research Groups (Grant No. 61721005), and Zhejiang University Education Foundation Global Partnership Fund. The National Supercomputer Center in Tianjin is acknowledged for computational support.

- Chinese Physics B的其它文章
- Helium bubble formation and evolution in NiMo-Y2O3 alloy under He ion irradiation
- Dynamics and intermittent stochastic stabilization of a rumor spreading model with guidance mechanism in heterogeneous network
- Spectroscopy and scattering matrices with nitrogen atom:Rydberg states and optical oscillator strengths
- Low-overhead fault-tolerant error correction scheme based on quantum stabilizer codes
- Transmembrane transport of multicomponent liposome-nanoparticles into giant vesicles
- Molecular dynamics simulations of A-DNA in bivalent metal ions salt solution