深度學(xué)習(xí)模型在中藥毒性預(yù)警中的應(yīng)用和前景
2022-04-13 02:50:22顏彩琴范睿琦寧雨坪
中國藥理學(xué)與毒理學(xué)雜志 2022年3期
顏彩琴,范睿琦,寧雨坪,郭 憲,王 凱
(1.天津中醫(yī)藥大學(xué)中藥學(xué)院,天津 301617;2.南開大學(xué)人工智能學(xué)院,天津 300350)
中藥潛在毒性成分的預(yù)警在中藥新藥研發(fā)、安全使用及風(fēng)險防控決策等方面具有重要意義。目前用于藥物毒性成分預(yù)警的方法和手段主要包括實驗方法和計算機預(yù)測方法。實驗方法主要是在實驗室中進(jìn)行動物實驗及體外細(xì)胞實驗,耗損率高,周期長,經(jīng)濟(jì)效益低[1]。計算機預(yù)測方法是利用機器學(xué)習(xí)模型直接對藥物毒性進(jìn)行預(yù)測,無需進(jìn)行大量實驗,經(jīng)濟(jì)效益高[2]。近年來,隨著訓(xùn)練數(shù)據(jù)的增多,機器學(xué)習(xí)技術(shù)得到快速發(fā)展。
早期用于毒性預(yù)警的機器學(xué)習(xí)方法是將藥物分子進(jìn)行編碼,然后輸入到傳統(tǒng)機器學(xué)習(xí)算法架構(gòu)的模型中,常見的有支持向量機(support vector machine,SVM)模型、隨機森林(random forest,RF)模型、樸素貝葉斯模型、決策樹模型和k-近鄰模型。傳統(tǒng)機器學(xué)習(xí)模型的優(yōu)點是需要的數(shù)據(jù)量少,模型訓(xùn)練快,然而其表征能力有限,預(yù)測準(zhǔn)確度較低[3]。隨著深度學(xué)習(xí)(deep learning,DL)算法,也稱為深度神經(jīng)網(wǎng)絡(luò)(deep neural network,DNN)的發(fā)展,DL模型逐漸替代傳統(tǒng)機器學(xué)習(xí)模型用于藥物毒性的預(yù)測[4]。與傳統(tǒng)機器學(xué)習(xí)模型相比,DL模型通過構(gòu)建多層人工神經(jīng)網(wǎng)絡(luò)(artificial neural network,ANN),從大量的數(shù)據(jù)中自動學(xué)習(xí)毒性化合物的結(jié)構(gòu)特征,其準(zhǔn)確性和可靠性得到極大提升。本文綜述了DL模型基于“結(jié)構(gòu)預(yù)警子-毒性”關(guān)系預(yù)測化合物潛在毒性的機制以及在藥物分子毒性預(yù)測和毒理學(xué)研究中的具體應(yīng)用,并提出DL模型在中藥毒性預(yù)警中的挑戰(zhàn)和展望,以期借助人工智能技術(shù)保障上市中藥臨床應(yīng)用的安全性。
1 中藥致毒機制及深度學(xué)習(xí)模型
1.1 “結(jié)構(gòu)預(yù)警子-毒性”關(guān)系
“結(jié)構(gòu)預(yù)警子-毒性”關(guān)系是機器學(xué)習(xí)模型預(yù)測化合物毒性的基本依據(jù)之一。通過識別數(shù)據(jù)集中的化合物結(jié)構(gòu)信息,機器學(xué)習(xí)模型能夠提取出化合物與毒性相關(guān)的結(jié)構(gòu)特征,從而預(yù)測目標(biāo)化合物的毒性[5]。
某些基團(tuán)被認(rèn)為是許多有毒中藥單體毒性的結(jié)構(gòu)標(biāo)志,如黃獨素B[6]、補骨脂素[7]和薄荷呋喃[8]中的呋喃環(huán),千里光堿[9]中的吡咯里西啶,馬錢子堿[10]、吳茱萸堿[11]和吳茱萸次堿[12]中的吲哚環(huán)及小檗紅堿[13]中的喹啉環(huán)等,這些基團(tuán)被稱為“結(jié)構(gòu)預(yù)警子”(structural alert)、毒理團(tuán)或毒性片段[14]。研究表明,具有“結(jié)構(gòu)預(yù)警子”的化合物引發(fā)毒性的關(guān)鍵一步是“結(jié)構(gòu)預(yù)警子”的氧化反應(yīng)[15-16]。在肝細(xì)胞色素P450酶的催化下,“結(jié)構(gòu)預(yù)警子”被氧化形成具有高度親電性的反應(yīng)性代謝產(chǎn)物(reactive metabolite,RM),這一過程被稱為化合物的“代謝活化”。RM可能與體內(nèi)重要生物大分子,如蛋白質(zhì)、酶和DNA的親核基團(tuán)發(fā)生共價結(jié)合,進(jìn)而引發(fā)一系列的毒性反應(yīng)。因此,在中藥研發(fā)早期,快速并全面篩查化合物中的“結(jié)構(gòu)預(yù)警子”并預(yù)測RM的形成是中藥毒理學(xué)研究的重要組成部分,對于規(guī)避中藥引發(fā)臨床毒性事件至關(guān)重要。
篩查化合物“結(jié)構(gòu)預(yù)警子”的實驗室方法,是在肝微粒體孵化體系中加入親核試劑,如谷胱甘肽、N-乙酰半胱氨酸和N-乙酰賴氨酸作為RM的捕獲劑,并借助液質(zhì)技術(shù),對捕獲劑共價結(jié)合RM所形成的穩(wěn)定加合物進(jìn)行定性,從而間接推斷出RM的結(jié)構(gòu)和原化合物發(fā)生代謝活化的反應(yīng)中心,即“結(jié)構(gòu)預(yù)警子”[17-18]。然而,實驗室方法耗時長且需要昂貴的檢測儀器,并且離不開人工操作,難以在短期內(nèi)實現(xiàn)大量化合物的檢測。為此,“結(jié)構(gòu)預(yù)警子”的概念自1985年被提出后,不同領(lǐng)域的專家將在自然界和合成化合物中發(fā)現(xiàn)的“結(jié)構(gòu)預(yù)警子”匯編形成一套“專家系統(tǒng)”,用于直觀、快速地評估化合物的毒性[19]。然而,由于基團(tuán)之間的相互影響,并非所有具有“結(jié)構(gòu)預(yù)警子”的化合物都發(fā)生代謝活化并致毒。所以,只根據(jù)“結(jié)構(gòu)預(yù)警子”預(yù)測化合物的毒性可能會出現(xiàn)假陽性結(jié)果,而且無法評估其毒性強弱[20]。
以DL模型為代表,近年發(fā)展起來的的機器學(xué)習(xí)模型能夠彌補“專家系統(tǒng)”方法的不足。事實上,兩者結(jié)合能獲得更準(zhǔn)確的預(yù)測結(jié)果。一方面,DL模型具有多層神經(jīng)網(wǎng)絡(luò),能夠綜合分析整個分子結(jié)構(gòu)而非局限于某個毒性基團(tuán),從而提取出超越“結(jié)構(gòu)預(yù)警子”的高層特征。另一方面,在建立數(shù)據(jù)集時,以“結(jié)構(gòu)預(yù)警子”作為輸入特征,能更好地訓(xùn)練DL模型對隱性特征的提取和組合能力[21-22]。因此,在藥物毒性預(yù)測中,與基于統(tǒng)計學(xué)的“專家系統(tǒng)”方法相比,基于“結(jié)構(gòu)預(yù)警子-毒性”關(guān)系的DL模型可能獲得更高的識別率。
1.2 深度學(xué)習(xí)模型的開發(fā)
在中藥的毒性預(yù)測中,DL模型的基本開發(fā)流程如圖1所示。首先是根據(jù)化合物的結(jié)構(gòu)或生物學(xué)特征對篩選出的化合物進(jìn)行分類,然后使用分子描述符將化合物的信息字符化,生成帶毒性標(biāo)簽的訓(xùn)練集并輸入到合適的DNN架構(gòu)中。DL模型通過逐層特征變換自動對化合物的數(shù)據(jù)進(jìn)行毒性表征,從而擁有預(yù)測化合物毒性的能力[23-24]。經(jīng)過驗證,若模型表現(xiàn)出較佳的性能,則可應(yīng)用于中藥毒性預(yù)警。
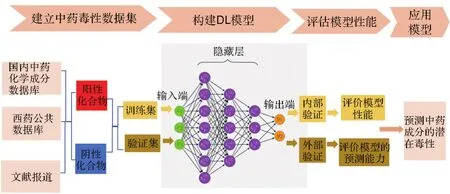
圖1 預(yù)測中藥毒性成分深度學(xué)習(xí)(DL)模型的開發(fā)流程[24].
1.2.1 毒性化合物數(shù)據(jù)集的建立
足夠數(shù)量和高質(zhì)量的數(shù)據(jù)是DL模型性能的保證[24],如數(shù)據(jù)量少且多樣性低下,則可能導(dǎo)致DL模型泛化不足,對數(shù)據(jù)產(chǎn)生偏向性。表1列舉了常用的毒性化合物數(shù)據(jù)庫。為保證數(shù)據(jù)集的質(zhì)量,在輸入模型之前,還需對采集的數(shù)據(jù)進(jìn)行篩選,過濾掉錯誤的以及重疊的數(shù)據(jù),并對化合物結(jié)構(gòu)進(jìn)行標(biāo)準(zhǔn)化。
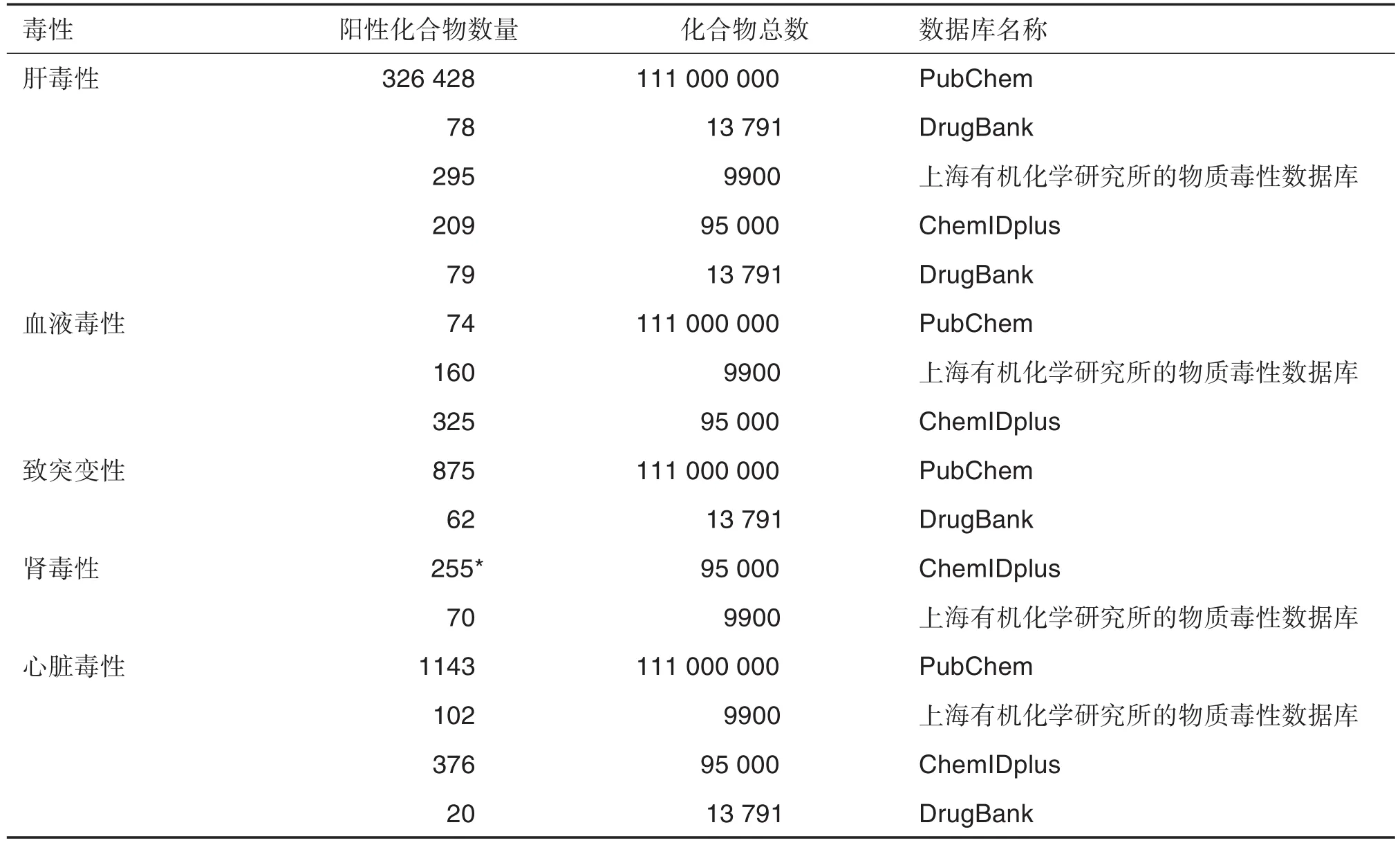
表1 常用毒性化合物數(shù)據(jù)庫
1.2.2 深度學(xué)習(xí)模型的構(gòu)建
在模型的構(gòu)建過程中,一般采用分子描述符將化合物的結(jié)構(gòu)和性質(zhì)編碼為模型能夠直接識讀和處理的數(shù)字化信息[25]。目前應(yīng)用于計算機預(yù)測的分子描述符數(shù)量達(dá)到幾百甚至上千,然而并無一種分子描述符普遍適用于編碼所有化學(xué)信息學(xué)數(shù)據(jù)[26]。因此,應(yīng)根據(jù)待解決的毒性預(yù)測問題和算法選擇合適的分子描述符。
分子指紋是一種特殊的分子描述符,常見的有擴(kuò)展聯(lián)通指紋、PubChem分子庫和分子接入系統(tǒng)等[27-29](表2)。分子指紋能將每一個分子的化學(xué)結(jié)構(gòu)簡化成獨特的字串符,用于表征內(nèi)部子結(jié)構(gòu)或官能團(tuán)是否存在,便于進(jìn)行2個相似化合物的比較[30]。

表2 常用分子指紋
當(dāng)前用于藥物毒性識別與預(yù)測的DL模型大體可以分為2類:深度前饋神經(jīng)網(wǎng)絡(luò)模型和深度圖神經(jīng)網(wǎng)絡(luò)模型。在深度前饋神經(jīng)網(wǎng)絡(luò)模型中,常用的深度神經(jīng)網(wǎng)絡(luò)模型是卷積神經(jīng)網(wǎng)絡(luò)(convolutional neural network,CNN)模型和循環(huán)或遞歸神經(jīng)網(wǎng)絡(luò)(recursive or recurrent neural network,RNN)模型。CNN模型是基于圖卷積的分子編碼方式,將輸入的原子水平信息組合為分子水平的信息,尤其擅長由低到高層次的學(xué)習(xí),從中提取出與任務(wù)最相關(guān)的一組特征[31]。在藥物設(shè)計與研發(fā)領(lǐng)域,常使用的CNN模型有基于神經(jīng)網(wǎng)絡(luò)的分子指紋模型、基于分子圖卷積(molecular graph convolution,MGC)模型和基于3D結(jié)構(gòu)的MGC模型,目前已被用于預(yù)測分子性質(zhì)和分子間的相互作用[32]。RNN是另一種前饋神經(jīng)網(wǎng)絡(luò),其單元之間的連接能夠形成環(huán),該特點使RNN模型能夠捕獲并反映動態(tài)中的短暫行為。RNN模型在醫(yī)學(xué)影像學(xué)中廣泛用于輔助診斷,近年來也被用于藥動學(xué)和藥效學(xué)的相關(guān)研究[33-34]。無向圖遞歸神經(jīng)網(wǎng)絡(luò)(undirected graph RNN,UGRNN)是RNN的一個新拓展,該方法將分子看作原子和鍵組成的無向圖,依次以每個重原子為根節(jié)點,將其他節(jié)點的信息沿最短路徑向根節(jié)點傳遞,每個根節(jié)點生成一個定長的向量,將所有定長向量相加得到分子水平的表征向量,輸入全連接網(wǎng)絡(luò)進(jìn)行決策[35]。UGRNN模型減少了對分子描述符的依賴,在自動學(xué)習(xí)中即可得到合適的數(shù)據(jù)表示,已應(yīng)用于預(yù)測化合物水溶性和藥物分子肝毒性,并獲得較高的準(zhǔn)確率[36]。
與前饋神經(jīng)網(wǎng)絡(luò)將分子描述符作為輸入不同,圖神經(jīng)網(wǎng)絡(luò)(graph neural network,GNN)模型直接基于圖對分子進(jìn)行編碼,其中分子中的原子作為圖的節(jié)點,原子與原子之間的鍵作為邊。GNN模型利用圖卷積操作實現(xiàn)信息在節(jié)點間的傳遞、可提取基團(tuán)等重要信息,在藥物毒性識別和可解釋性方面GNN模型具有明顯優(yōu)勢。另外,GNN又可與CNN、RNN和注意力機制等聯(lián)合使用形成多種深度GNN模型,如圖注意網(wǎng)絡(luò)模型、圖卷積網(wǎng)絡(luò)模型[37]和多任務(wù)圖注意模型[38]等,這些模型在藥物毒性識別中取得當(dāng)前最好的結(jié)果。GNN模型、CNN模型和RNN模型的優(yōu)缺點分析見表3。

表3 圖神經(jīng)網(wǎng)絡(luò)、卷積神經(jīng)網(wǎng)絡(luò)和循環(huán)神經(jīng)網(wǎng)絡(luò)的優(yōu)缺點
1.2.3 深度學(xué)習(xí)模型的驗證與評估
模型生成后,下一步需對模型的擬合能力、穩(wěn)定性以及預(yù)測能力進(jìn)行驗證與評價。驗證包括內(nèi)部驗證和外部驗證,分別用于評價模型的穩(wěn)定性和模型的預(yù)測能力[37]。內(nèi)部驗證通過多次迭代和對測試集上的性能進(jìn)行平均,可以減小隨機分割數(shù)據(jù)集帶來的偶然因素的影響,常見的方法是蒙特卡羅交叉驗證[39]和 k折交叉驗證[40]。外部驗證使用獨立于訓(xùn)練集的數(shù)據(jù)測試模型的泛化性,并檢查是否出現(xiàn)“過擬合”問題[41]。
在模型性能評價中,常使用的指標(biāo)包括靈敏性(sensitivity,SE)、特異性(specificity,SP)、準(zhǔn)確性(accuracy,ACC)、精確率(precision,P)、ROC曲線下面積(area under the receiver operating characteristic curve,AUC)和 PR 曲線下面積(area under the precision recall curve,AUPR)[42]。在DL模型對藥物毒性的預(yù)測結(jié)果中,SE即真陽性率,指所有毒性化合物中被正確預(yù)測為陽性的比例;SP即真陰性率,指所有無毒化合物中被正確地預(yù)測為陰性的比例;ACC即被正確預(yù)測為有毒的陽性化合物和被正確預(yù)測為無毒的陰性化合物占所有化合物的比例,準(zhǔn)確率越高,模型越好;P為真毒性化合物數(shù)量占陽性結(jié)果數(shù)量的比例。
另外,ROC曲線是以假陽性率為橫軸,SE為縱軸繪制的曲線。AUC值指隨機給定一個有毒性化合物和一個無毒性化合物,模型輸出有毒性化合物為陽性的概率值比輸出無毒性化合物為陰性的概率值大的可能性。當(dāng)AUC=0.5時,真陽性率和假陽性率相等,相當(dāng)于隨機猜測,AUC趨于1,即ROC曲線越靠近左上方時,模型的預(yù)測性能越好。與ROC曲線不同的是,PR曲線是以SE為橫軸,以P為縱軸繪制的曲線。PR曲線越靠近右上方,表示模型的預(yù)測性能越好。
2 深度學(xué)習(xí)模型在藥物毒性預(yù)測和毒理學(xué)研究中的應(yīng)用
基于化學(xué)結(jié)構(gòu)與毒性的關(guān)系,一般是使用已知的陽性化合物和陰性化合物的結(jié)構(gòu)信息作為數(shù)據(jù)集進(jìn)行模型訓(xùn)練和驗證,使模型能夠識別毒性化合物的結(jié)構(gòu)特征。目前,DL模型已作為藥物毒理學(xué)研究的工具,被用于解決藥物分子及其代謝產(chǎn)物的毒性預(yù)測問題。然而,在藥物毒理學(xué)研究中,除篩查出更多有潛在危害性的藥物分子,還需進(jìn)一步明確其致毒的生物學(xué)靶標(biāo)和機制。因此,最近研發(fā)的DL模型同時把化學(xué)結(jié)構(gòu)信息和生物學(xué)數(shù)據(jù)輸入到模型中,使模型通過深度挖掘毒性藥物分子的化學(xué)生物學(xué)數(shù)據(jù)以發(fā)現(xiàn)新的規(guī)律[43]。
2.1 預(yù)測藥物分子的毒性
Xu等[36]使用NCTR數(shù)據(jù)集、Greene數(shù)據(jù)集、Xu數(shù)據(jù)集以及一個混合數(shù)據(jù)集的2788個化合物數(shù)據(jù)作為訓(xùn)練集,利用UGRNN分子編碼方法構(gòu)建藥物肝損傷預(yù)測模型。在外部驗證中,該模型的各性能指標(biāo)均高于淺層神經(jīng)網(wǎng)絡(luò)架構(gòu)的模型。2018年,Ambe等[44]針對肝細(xì)胞增生,在ORAD數(shù)據(jù)集、HESS數(shù)據(jù)集和一個混合數(shù)據(jù)集中比較3種機器學(xué)習(xí)算法,DNN,SVM,與RF預(yù)測化合物肝毒性的表現(xiàn)。經(jīng)過驗證,使用數(shù)據(jù)量較小的HESS數(shù)據(jù)集和SVM算法構(gòu)建的模型獲得最佳的預(yù)測性能。由此可見,在實際應(yīng)用中,DL模型并不總是優(yōu)于傳統(tǒng)機器學(xué)習(xí)模型,這可能與數(shù)據(jù)集的大小有關(guān)。一般而言,傳統(tǒng)機器學(xué)習(xí)模型適合處理規(guī)模較小的數(shù)據(jù)集,DL模型則在分析多且復(fù)雜的數(shù)據(jù)中顯示出杰出的優(yōu)勢[45]。
類似地,基于化學(xué)結(jié)構(gòu)的方法,Hua等[20]針對藥物分子的血液毒性,使用從SIDER數(shù)據(jù)庫、ChemIDplus數(shù)據(jù)庫以及文獻(xiàn)數(shù)據(jù)中篩選出的632個陽性化合物和1525個陰性化合物作為數(shù)據(jù)集,在未采用分子描述符的情況下直接將整個分子輸入算法中,構(gòu)建了預(yù)測藥物分子血液毒性的CNN模型。在外部驗證中,該模型ACC為76%,AUC為64%,SP為81%。由此可見,相較于傳統(tǒng)機器學(xué)習(xí)模型,DL模型的另一個優(yōu)勢在于不依賴分子描述符編碼,能將原始的輸入信息進(jìn)行轉(zhuǎn)換。
2021年,Kumar等[46]使用從現(xiàn)有文獻(xiàn)中篩選出的4053種化合物作為數(shù)據(jù)集,比較DNN,SVM,RF和k-近鄰幾種算法建立的模型在預(yù)測藥物分子致突變性中的表現(xiàn)。通過測試,DNN模型在測試集和驗證集中均獲得最高的ACC,分別為95.0%和83.8%。表4總結(jié)了近年來用于藥物毒性預(yù)測的DL模型。根據(jù)模型在外部驗證集的ACC和AUC針對不同的藥物毒性預(yù)測問題,DL模型均具有較高的預(yù)測能力。
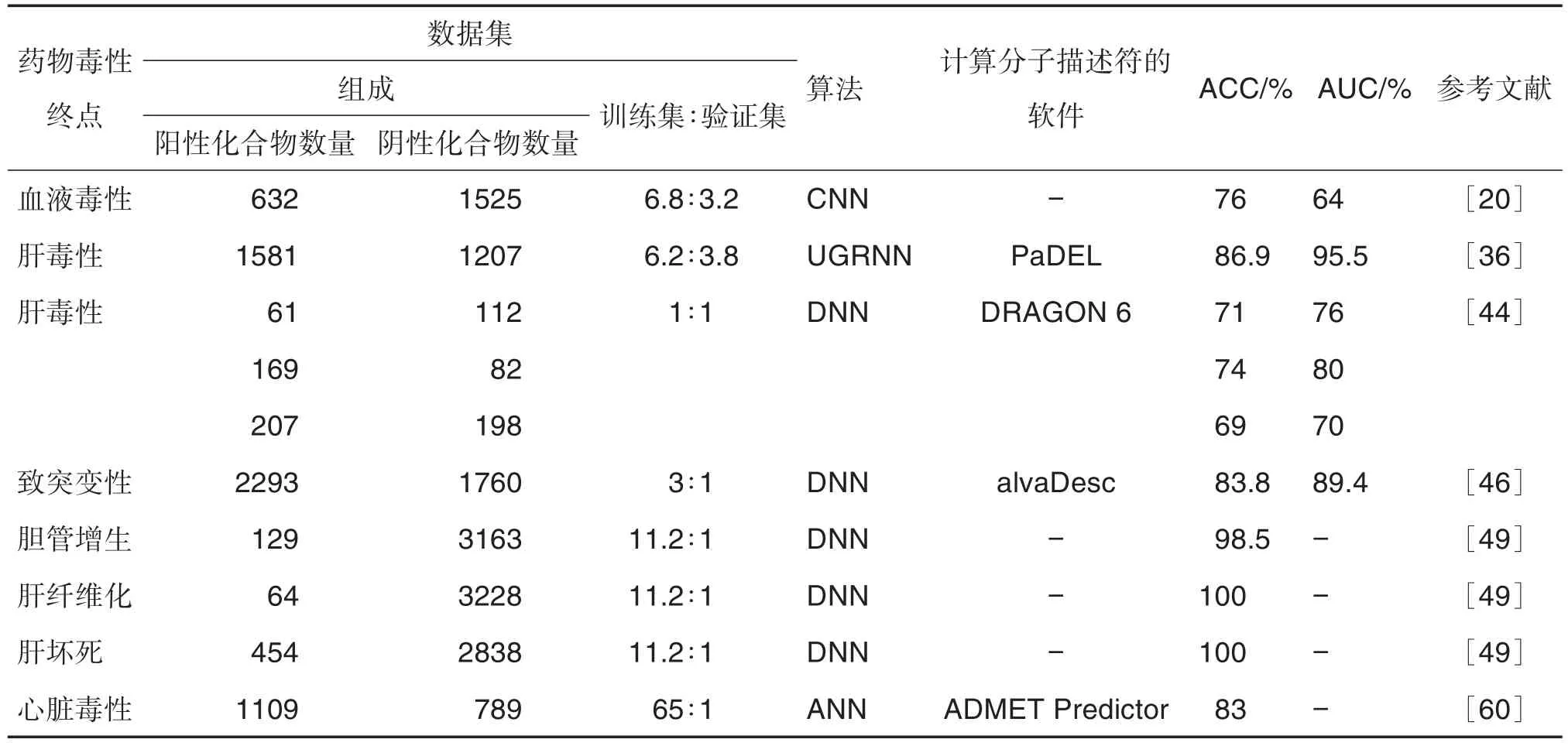
表4 深度學(xué)習(xí)模型預(yù)測藥物不同毒性終點的性能
2.2 預(yù)測毒性反應(yīng)性代謝產(chǎn)物的形成
2017年,Hughes等[47]首次構(gòu)建出預(yù)測化合物經(jīng)代謝活化形成醌類RM的CNN模型。他們在CNN架構(gòu)中設(shè)置了1個輸入層,3個隱藏層及3個輸出層。隱藏層從低到高分別為原子層、原子對層、分子層,代表了藥物分子在3個水平上形成醌類RM的能力。該CNN模型通過在0~1的范圍內(nèi)給每一層打分,以識別出化合物最有可能發(fā)生代謝活化的位點,并且綜合評估化合物分子形成醌類RM的能力。在最近的一項研究中,Wang等[48]使用DL算法構(gòu)建出預(yù)測藥物分子的代謝反應(yīng)類型及其潛在危害性代謝產(chǎn)物的機器學(xué)習(xí)模型。在藥物毒理學(xué)的體內(nèi)、體外研究中,DL模型能結(jié)合分子背景,較全面、準(zhǔn)確、快速地預(yù)測出化合物經(jīng)過轉(zhuǎn)化形成毒性代謝產(chǎn)物而致毒的可能性,減少了實驗的次數(shù)和周期。
2.3 預(yù)測藥物分子致毒的生物學(xué)靶標(biāo)
生物學(xué)數(shù)據(jù)集與DL模型結(jié)合的優(yōu)勢,一方面在于生物學(xué)數(shù)據(jù)涵蓋從單個基因表達(dá)到多個基因富集通路上的分子水平信息,可以為模型提供成千上萬的輸入變量,DL模型通過增加隱藏層的數(shù)目能在同一時間對輸入的變量進(jìn)行學(xué)習(xí),從而提高特征泛化能力;另一方面,在訓(xùn)練集中增加毒性相關(guān)基因和途徑的生物學(xué)信息,利用DL模型的特征識別和提取能力,有助于發(fā)現(xiàn)藥物分子致毒的生物學(xué)靶點和機制[43]。Wang等[49]針對膽管增生、肝纖維化和肝壞死3個肝毒性終端,使用DrugMatrix和OpenTG-GATEs數(shù)據(jù)庫的9876個樣本的毒理基因組學(xué)數(shù)據(jù)作為數(shù)據(jù)集,建立了預(yù)測肝毒性的DNN模型。該模型具有多個隱藏層,并且輸入端和隱藏層均包含多個神經(jīng)元,通過多次非線性轉(zhuǎn)換可學(xué)習(xí)到更加復(fù)雜和抽象的特征。結(jié)果顯示,在3種毒性終點基因和相關(guān)通路的識別中,與SVM和RF相比,采用DNN建立的模型顯示出更好的穩(wěn)定性和特異性。
3 深度學(xué)習(xí)模型在中藥毒性預(yù)警中的挑戰(zhàn)和展望
DL模型將有可能成為解決中藥毒性預(yù)測這一難題的最優(yōu)選擇。目前,DL模型在中藥研發(fā)領(lǐng)域方興未艾,但同時面臨一些瓶頸問題。
3.1 中藥單體的毒性數(shù)據(jù)較少
DL模型要在中藥成分的毒理學(xué)研究中發(fā)揮巨大效用,建立高質(zhì)量的數(shù)據(jù)集是首要前提[50-51]。有研究表明,與僅由西藥化合物構(gòu)成的數(shù)據(jù)集訓(xùn)練出的模型相比,中藥成分參與構(gòu)成的混合數(shù)據(jù)庫完善了訓(xùn)練集的化學(xué)結(jié)構(gòu)空間,并有效降低機器學(xué)習(xí)模型的預(yù)測偏好性,提高了結(jié)果的準(zhǔn)確性,因而更適用于中藥成分的安全性評價[52]。然而,目前對于中藥中具有潛在毒性的單體成分的研究相對較少,在國內(nèi)外公共藥物數(shù)據(jù)庫中,中藥單體的毒理學(xué)數(shù)據(jù)更是寥寥無幾[53]。用于訓(xùn)練的毒理學(xué)數(shù)據(jù)有限,有可能使得DL模型在學(xué)習(xí)過程中泛化不全面,所學(xué)習(xí)到的毒性團(tuán)僅限于數(shù)據(jù)集中的化合物。另外,通過文獻(xiàn)檢索得到的中藥單體數(shù)據(jù)質(zhì)量往往參差不齊。很多文獻(xiàn)報道的因服用中藥而引發(fā)的臨床毒性事件中,無法排除醫(yī)生辨證用藥正確與否、患者是否誤用濫用以及有無基礎(chǔ)疾病等因素的影響,因此難以對檢索到的中藥單體進(jìn)行正確的標(biāo)記與分類[54]。
為獲得高質(zhì)量的數(shù)據(jù)集,建模前需要毒理學(xué)專家結(jié)合臨床使用劑量和機體的復(fù)雜性進(jìn)行全面分析,對數(shù)據(jù)進(jìn)行嚴(yán)格的篩選、標(biāo)注和分類。最根本的是要重視具有潛在毒性的中藥單體“結(jié)構(gòu)預(yù)警子”方面的深入研究,不斷積累相關(guān)的實驗數(shù)據(jù),從源頭上解決中藥單體毒理學(xué)數(shù)據(jù)較少的問題。
3.2 深度學(xué)習(xí)模型的黑箱問題
由于DL架構(gòu)過程是端到端,由輸入端到輸出端的因果關(guān)系的可解釋性是DL模型落地于中藥毒性預(yù)測必需的“通行證”[55-56]。為此,可以從2方面完善DL模型的可解釋性。一方面,從預(yù)測原理方面入手,結(jié)合“結(jié)構(gòu)預(yù)警子-毒性”關(guān)系和生物學(xué)致毒機制解釋模型的決策依據(jù),提高預(yù)測結(jié)果的可信度;另一方面,從模型的架構(gòu)入手,通過調(diào)試函數(shù)對模型的架構(gòu)造成微小的擾動,以確定不同變量對模型的影響,便于理解模型的行為和改進(jìn)模型[57-58]。
3.3 深度學(xué)習(xí)模型在中藥毒性預(yù)警中的展望
借助于集成式建模軟件,如ADMET Predictor和Virtual Tox Lab,機器學(xué)習(xí)模型已投入到中藥毒理學(xué)研究并展現(xiàn)出良好的預(yù)測性能[59-60]。如李雅秋等[60]針對心臟毒性使用ToxRefDB數(shù)據(jù)庫和SIDER數(shù)據(jù)庫中1109個陽性化合物和789個陰性化合物作為訓(xùn)練集,另外使用29種已知毒性的中藥成分組成外部驗證集,并采用ADMET Predictor軟件中自帶的SVM和ANN算法模塊構(gòu)建模型。在外部驗證集中,ANN模型預(yù)測的SE為95%,SP為60%,ACC為83%,明顯優(yōu)于SVM模型。
He等[61]使用由檢索科學(xué)出版物得到的296個肝毒性中藥成分和584個無肝毒性中藥成分以及公開的CTD數(shù)據(jù)庫組成的數(shù)據(jù)集,利用多種傳統(tǒng)機器學(xué)習(xí)算法和分子描述符構(gòu)建了數(shù)個基本分類器,將其中預(yù)測性能最佳的基本分類器進(jìn)行組合,建立了一款預(yù)測中藥肝毒性的模型。經(jīng)檢驗,該組合型預(yù)測模型在交叉驗證和外部驗證中均表現(xiàn)出優(yōu)于單個分類器的預(yù)測性能。值得一提的是,雖然組合式學(xué)習(xí)能夠通過增加基本分類器的數(shù)目提高傳統(tǒng)機器學(xué)習(xí)算法處理復(fù)雜數(shù)據(jù)的能力,但各基本分類器的預(yù)測性能不一,組合后不一定能獲得優(yōu)勢互補的效果。迄今為止,DL模型在中藥成分毒性評價中的應(yīng)用還比較少。
總體來看,傳統(tǒng)機器學(xué)習(xí)模型高度依賴人工設(shè)計的輸入特征,普適性低且易出現(xiàn)“過擬合”;DL模型處理分析大數(shù)據(jù)和復(fù)雜數(shù)據(jù)更勝一籌。針對中藥多組分、多靶標(biāo)的特點,DL模型有能力深度挖掘出中藥毒性成分的結(jié)構(gòu)和生物學(xué)特征。若在中藥研發(fā)早期利用DL模型實現(xiàn)高效、快速的風(fēng)險預(yù)警,既能把毒性大的中藥成分及時剔除,也能為具有潛在危害性的藥物候選分子的結(jié)構(gòu)優(yōu)化提供理論指導(dǎo)[20]。
4 結(jié)語
中藥成分復(fù)雜,篩查具有潛在毒性的中藥成分并揭示其致毒機制是當(dāng)前亟待解決的難題。在藥物的毒性評價中,與耗損率高、周期長的實驗室方法相比,DL模型幾乎不需要成本、人力和物力,即可快速、準(zhǔn)確地給出預(yù)測結(jié)果。與傳統(tǒng)機器學(xué)習(xí)模型相比,DL模型在處理和分析復(fù)雜體系的信息上更具優(yōu)勢。雖然目前在數(shù)據(jù)來源和模型可解釋性方面仍有不足,但DL模型在中藥毒性預(yù)警中的廣闊應(yīng)用前景不可否認(rèn)。隨著DL模型的不斷優(yōu)化以及更多潛在毒性中藥成分被揭示和積累成規(guī)范的數(shù)據(jù)庫,相信在不久后,DL模型將突破現(xiàn)有的瓶頸,成為新一代預(yù)測中藥潛在毒性的杰出工具。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
中老年保健(2021年4期)2021-12-01 11:19:40
中老年保健(2021年4期)2021-08-22 07:08:32
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年10期)2020-11-26 08:24:50
金橋(2020年7期)2020-08-13 03:07:00
基層中醫(yī)藥(2020年12期)2020-07-22 06:34:38
數(shù)學(xué)物理學(xué)報(2020年2期)2020-06-02 11:29:24
基層中醫(yī)藥(2018年6期)2018-08-29 01:20:20
光學(xué)精密工程(2016年6期)2016-11-07 09:07:19
核科學(xué)與工程(2015年4期)2015-09-26 11:59:03
