高效液相色譜法測定含對乙酰氨基酚、氫溴酸右美沙芬和琥珀酸多西拉敏的軟膠囊有關物質
2022-04-19 00:39:10邱科先劉曉芬
中國藥物經濟學 2022年3期
邱科先 劉曉芬
感冒作為一種常見的呼吸道疾病,其主要臨床表現為頭疼、發熱、咳嗽、鼻塞、流鼻涕、過敏等。通常用于治療這些癥狀的藥物包括解熱鎮痛藥(對乙酰氨基酚)、鎮咳藥(氫溴酸右美沙芬)、抗組胺藥(琥珀酸多西拉敏)[1-4]。美國Vicks的感冒藥有DayQuil和NyQuil兩種,其中NyQuil的主成分是對乙酰氨基酚、琥珀酸多西拉敏和氫溴酸右美沙芬。據此,采用明膠制成一種含對乙酰氨基酚、氫溴酸右美沙芬和琥珀酸多西拉敏的軟膠囊的復方口服固體制劑,其制備方法如中國專利201310359375.2所述[5],目前對于該產品的研究仍有待改進。
美國藥典43版中,琥珀酸多西拉敏的有關物質測定采用高效液相色譜法(HPLC)[6];《中華人民共和國藥典》2020年二部中,對乙酰氨基酚和氫溴酸右美沙芬的有關物質測定采用HPLC[7-8],但色譜條件不同。現有技術中還沒有能夠同時測定含對乙酰氨基酚、氫溴酸右美沙芬和琥珀酸多西拉敏的軟膠囊有關物質的方法[6-10]。為節約時間和人力,方便生產檢驗,將上述3種物質的有關物質測定方法進行統一,本研究采用同一種色譜條件測定含對乙酰氨基酚、氫溴酸右美沙芬和琥珀酸多西拉敏的軟膠囊有關物質,并進行方法驗證。
1 材料與方法
1.1 藥品與試劑
一種含對乙酰氨基酚、氫溴酸右美沙芬和琥珀酸多西拉敏的軟膠囊(規格對乙酰氨基酚325 mg;琥珀酸多西拉敏6.25 mg;氫溴酸右美沙芬15 mg);對乙酰氨基酚內部對照品(批號RS-18099,純度99.0%)購自安丘市魯安藥業有限責任公司;氫溴酸右美沙芬(批號100201-201204,純度94.8%)購自中國食品藥品檢定研究院;琥珀酸多西拉敏(批號R08200,純度99.9%,USP對照品)購自赫淳生物科技(上海)有限公司;右美沙芬雜質Ⅰ(批號38400,純度99.7%)、右美沙芬雜質Ⅱ(批號145192,純度98.7%)、右美沙芬雜質Ⅲ(批號35952,純度99.7%)、右美沙芬雜質Ⅳ(批號32441,純度98.1%)購自英國LGC公司。
1.2 儀器
Agilent 1260型高效液相色譜儀(美國Agilent-Technologies公司)和Thermo Fisher Ultimate3000型高效液相色譜儀(賽默飛世尓科技有限公司),包括在線脫氣機、四元泵、自動進樣器、二極管陣列檢測器;電子天平Secura255D、酸度計(賽多利斯科學儀器(北京)有限公司);超聲波清洗儀(深圳德瑞超聲波設備有限公司);多功能振蕩器(常州國華電器有限公司);電熱恒溫鼓風干燥箱(上海一恒科學儀器有限公司);藥品強光穩定性試驗箱(北京蘭貝石恒溫技術有限公司)
1.3 方法
1.3.1 溶液的制備 1)稀釋液:0.01 mol/L鹽酸-甲醇(8∶2)。2)雜質對照品溶液:取右美沙芬雜質Ⅰ、右美沙芬雜質Ⅱ、右美沙芬雜質Ⅲ、右美沙芬雜質Ⅳ對照品各適量,精密稱定,用稀釋液溶解并稀釋,制成含各已知雜質為40 μg/ml的混合溶液,作為雜質對照儲備溶液;取上述雜質對照儲備溶液2 ml,用稀釋液稀釋至20 ml,作為雜質對照品溶液。3)工作對照溶液:取對乙酰氨基酚、琥珀酸多西拉敏和氫溴酸右美沙芬對照品適量,精密稱定,用稀釋液溶解并稀釋,制成含對乙酰氨基酚91 μg/ml、琥珀酸多西拉敏1.75 μg/ml和氫溴酸右美沙芬4.2 μg/ml的溶液,作為工作對照溶液。4)供試品溶液:取含對乙酰氨基酚、氫溴酸右美沙芬和琥珀酸多西拉敏的軟膠囊,置于250 ml量瓶中,加約120 ml稀釋液,加熱使囊殼崩解,震搖使混合均勻,用稀釋液稀釋,制成含對乙酰氨基酚9 100 μg/ml、琥珀酸多西拉敏175 μg/ml和氫溴酸右美沙芬420 μg/ml的溶液,濾過,作為供試品溶液。5)自身對照溶液:取“4)”項下供試品溶液1 ml,置于100 ml量瓶中,用稀釋液稀釋至刻度,搖勻,作為自身對照溶液。6)系統適用性溶液:取“2)”項下雜質對照儲備溶液1 ml,置于10 ml量瓶中,用“3)”項下工作對照溶液稀釋至刻度,搖勻,作為系統適用性溶液。
1.3.2 色譜條件 色譜柱:Phenomenex Luna C18(250×4.6 mm,5 μm);流動相A:多庫酯鈉和乙酸銨的水溶液(取多庫酯鈉4.45 g和乙酸銨1.54 g,加水2 000 ml,溶解,調節pH至4.5);流動相B:乙腈-冰醋酸(2∶1),梯度洗脫見表1;流速:1.0 ml/min;柱溫:30 ℃;檢測波長:280 nm;進樣量:50 μl。

表1 梯度洗脫程序
1.3.3 系統適用性試驗 取系統適用性溶液50 μl,按“1.3.2”項下色譜條件進樣測定,記錄色譜圖。
1.3.4 強降解試驗 取含對乙酰氨基酚、氫溴酸右美沙芬和琥珀酸多西拉敏的軟膠囊適量,置于一次性塑料培養皿,分別置于高溫(60 ℃烘箱12 d),光照(4 500 Lux和400 μW/cm2,12 d)條件下進行強制破壞,破壞結束后按照供試品溶液的配制方式配制溶液;取含對乙酰氨基酚、氫溴酸右美沙芬和琥珀酸多西拉敏的軟膠囊適量,置于100 ml量瓶中,分別酸降解(加0.5 mol/L鹽酸溶液5 ml,60 ℃水浴中放置2 h)、堿降解(加0.5 mol/L氫氧化鈉溶液5 ml,60 ℃水浴中放置2 h)、氧化降解(加入20 ml稀釋液置于50 ℃水浴中加熱使囊殼崩解,加30%過氧化氫溶液室溫放置2 h),破壞后取出溶液,冷卻至室溫,堿降解溶液通過加0.5 mol/L鹽酸溶液5 ml中和溶液,酸降解溶液通過加0.5 mol/L氫氧化鈉溶液5 ml中和溶液,再分別用稀釋液稀釋至刻度,濾過,取濾液按“1.3.2”項下色譜條件進樣測定,記錄色譜圖。
1.3.5 線性研究 取對乙酰氨基酚、琥珀酸多西拉敏、氫溴酸右美沙芬和各已知雜質對照品適量,精密稱定,加稀釋液稀釋,制成各成分濃度相對于樣品中主成分濃度0.05%、0.25%、0.50%、1.00%和2.00%水平的溶液,作為線性溶液。取上述溶液按“1.3.2”項下色譜條件進樣測定,記錄峰面積。以峰面積為縱坐標(y)、進樣質量濃度為橫坐標(x)進行線性回歸,獲得相關系數和計算相對響應因子。
1.3.6 檢測限和定量限 取各雜質線性溶液的最低濃度溶液,用稀釋液稀釋成一系列溶液,按“1.3.2”項下色譜條件進樣測定,記錄色譜圖。以能檢測的最低濃度作為檢測限濃度,以能定量的最低濃度作為定量限濃度。
1.3.7 精密度試驗 1)重復性 取含對乙酰氨基酚、氫溴酸右美沙芬和琥珀酸多西拉敏的軟膠囊,置于250 ml量瓶中,加入相對于樣品溶液的0.28%水平的雜質,加約150 ml稀釋液,加熱使囊殼崩解,震搖使混合均勻,用稀釋液稀釋,濾過,配制6份,按“1.3.2”項下色譜條件進樣測定,記錄色譜圖。采用加校正因子的主成分自身對照法計算雜質的含量(未知雜質校正因子按1.0計算),采用不加校正因子的主成分自身對照法計算未知雜質的含量。2)中間精密度 由另一試驗員于不同測試日期按重復性項下配制6份溶液,用不同的HPLC系統,不同的對照溶液和不同批號的柱子,按“1.3.2”項下色譜條件進樣測定,記錄色譜圖,并按重復性項下計算各雜質的含量。
1.3.8 加樣回收率試驗 取含對乙酰氨基酚、氫溴酸右美沙芬和琥珀酸多西拉敏的軟膠囊,置于250 ml量瓶中,分別加入相對于樣品溶液的0.05%、0.24%、0.95%和1.43%水平的雜質,加約150 ml稀釋液,加熱使囊殼崩解,震搖使混合均勻,用稀釋液稀釋,濾過,每個水平配制3份,按“1.3.2”項下色譜條件進樣測定,記錄色譜圖。采用加校正因子的主成分自身對照法計算各已知雜質的測得濃度,再計算回收率。
1.3.9 穩定性試驗 將“1.3.7”項下重復性樣品溶液分別放置室溫條件和冰箱(2~8 ℃)條件下儲存4 d,4 d后按“1.3.2”項下色譜條件進樣測定,記錄色譜圖。
1.3.10 耐用性試驗 1)改變流動相初始比例 按“1.3.7”項下重復性樣品溶液的配制方式配制一份樣品溶液,按“1.3.2”項下條件以不同流動相初始比例(46∶54、48∶52)進樣,記錄峰面積,并按“1.3.7”項下計算各雜質的含量。2)改變流動相A的pH“1.3.7”項下重復性樣品溶液的配制方式配制一份樣品溶液,按“1.3.2”項下條件以不同流動相A pH(4.45、4.55)進樣,記錄峰面積,并按“1.3.7”項下計算各雜質的含量。
2 結果
2.1 系統適用性試驗
結果表明系統適用性溶液中,各已知雜質間的分離度均大于1.5。見圖1。
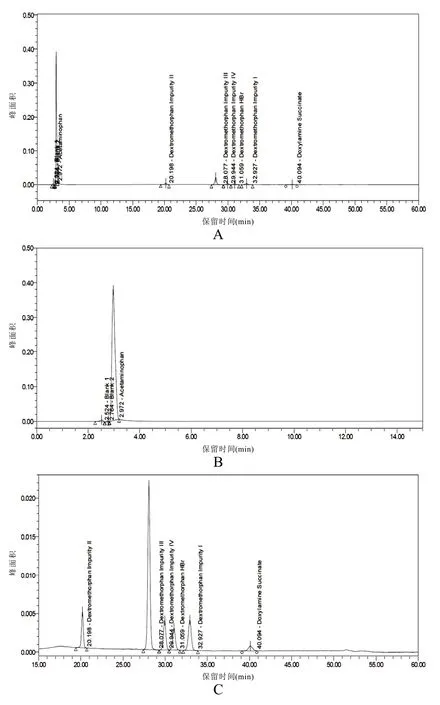
圖1 系統適用性溶液的高效液相色譜
2.2 強降解試驗
結果表明,在上述破壞條件下,各主成分的純度角均小于純度閾值,對乙酰氨基酚與最近的峰的分離度在1.0~1.4,氫溴酸右美沙芬與最近的峰的分離度均為2.4,琥珀酸多西拉敏與最近的峰的分離度在8.2~8.5。各已知雜質、未知雜質與主成分的分離度均大于1.0,均能較好分離。
2.3 線性研究
結果顯示各待測成分的相關系數均為1.00,右美沙芬雜質Ⅰ、右美沙芬雜質Ⅱ、右美沙芬雜質Ⅲ和右美沙芬雜質Ⅳ相對于右美沙芬的相對響應因子分別為1.22、0.79、4.99和1.05。
2.4 檢測限和定量限
結果表明主成分和各雜質的檢測限在0.000 2%~0.024%范圍內,定量限在0.000 4%~0.043%范圍內,低于0.05%。
2.5 精密度試驗
結果表明,重復性和中間精密度的相對標準偏差(RSD)在0%~8%范圍內,均小于10%。
2.6 加樣回收率試驗
結果表明,右美沙芬雜質Ⅰ、右美沙芬雜質Ⅱ、右美沙芬雜質Ⅲ和右美沙芬雜質Ⅳ的平均回收率在91%~114%范圍內,RSD均小于10%。
2.7 穩定性試驗
結果表明,各已知雜質的差值在0.01%~0.08%范圍內,對乙酰氨基酚總雜質的差值均為0.01%,均未檢出琥珀酸多西拉敏雜質,氫溴酸右美沙芬總雜質的差值在0.1%~0.2%范圍內。
2.8 耐用性試驗
結果表明,在色譜條件輕微變動情況下,各已知雜質的差值在0.01%~0.02%范圍內,對乙酰氨基酚總雜質的差值為0.00%~0.01%,均未檢出琥珀酸多西拉敏雜質,氫溴酸右美沙芬總雜質的差值在0.02%~0.03%范圍內。
3 討論
本研究所建方法擬檢測的是含對乙酰氨基酚、氫溴酸右美沙芬和琥珀酸多西拉敏的軟膠囊中對乙酰氨基酚有關物質、氫溴酸右美沙芬有關物質(右美沙芬雜質Ⅰ、右美沙芬雜質Ⅱ、右美沙芬雜質Ⅲ、右美沙芬雜質Ⅳ和未知雜質)、琥珀酸多西拉敏有關物質。通過驗證試驗,結果表明該色譜條件下能有效分離各已知雜質和主成分,上述色譜條件對雜質的測定無影響,同時本方法能滿足雜質檢測靈敏度的要求,精密度和準確度均良好。此外,樣品溶液在室溫條件和冰箱(2~8 ℃)條件可穩定儲存至少4 d,本方法在改變一定流動相初始比例、流動相A的pH變動情況下耐用性良好。
通過原輔料和成品的強降解研究,確認未知雜質是輔料峰或是來源于各主成分的有關物質,進而采用HPLC峰保留時間對比法進行各個主成分的有關物質的定性歸屬[11]。
對乙酰氨基酚降解會產生雜質——對氨基酚,因帶有苯胺結構,具有潛在的基因毒性。本研究色譜采集時間長,而對氨基酚需避光,且穩定性較其他雜質差,因此不采用本研究方法進行對氨基酚的測定。
3.1 測定波長的選擇
對乙酰氨基酚、琥珀酸多西拉敏、氫溴酸右美沙芬和右美沙芬雜質Ⅰ、右美沙芬雜質Ⅱ、右美沙芬雜質Ⅲ、右美沙芬雜質Ⅳ對照品,以“1.3.1”項下稀釋液作為溶劑進行紫外光譜掃描,氫溴酸右美沙芬、右美沙芬雜質Ⅰ、右美沙芬雜質Ⅳ結果在280 nm的波長處有1個較強的吸收峰,其他雜質和主成分也有較大吸收,因此選用280 nm作為有關物質的測定波長。
3.2 流動相水相的選擇
在本研究中,以不同pH的多庫酯鈉和乙酸銨的水溶液(pH為4.3~4.7)分別作為流動相水相,按照一定比例組成流動相梯度洗脫,從各主成分及其有關物質的分離度、拖尾因子和信噪比綜合考慮,選擇pH 4.5的多庫酯鈉和乙酸銨的水溶液作為流動相水相[12]。
3.3 洗脫方式的選擇
在本研究中,以“1.3.2”項下色譜條件中流動相A∶流動相B不同比例的流動相進行梯度洗脫,從洗脫時間、各雜質的分離情況、各主成分和各雜質的信噪比、基線平穩情況等方面綜合考慮,最終確定了有關物質的梯度洗脫方式[12]。
3.4 進樣體積的選擇
在本研究中,以25 μl和50 μl分別作為進樣體積,從各雜質的分離情況、各主成分和各雜質的信噪比等方面綜合考慮,最終確定了50 μl作為進樣體積[12]。
3.5 柱溫箱溫度的選擇
在本研究中,以25 ℃、30℃、35℃分別作為柱溫,從各雜質的分離情況、主成分的拖尾因子和運行時間等綜合考慮,最終確定了30 ℃作為柱溫箱的溫度[12]。
綜上所述,本研究所建方法能同時測定對乙酰氨基酚、琥珀酸多西拉敏和氫溴酸右美沙芬有關物質,簡便高效,靈敏度高,準確度、精密度、穩定性和耐用性良好。各已知雜質、未知雜質能與主成分和輔料峰有效分離,可用于含對乙酰氨基酚、氫溴酸右美沙芬和琥珀酸多西拉敏的軟膠囊有關物質的質量控制。
