斑翅果蠅YolkProtein1基因原核表達及抗體制備
2022-04-27 12:59:04祝晴王瑞娟覃冬云陳浩代曉彥劉艷周紫章翟一凡
山東農業科學 2022年3期
祝晴 王瑞娟 覃冬云 陳浩 代曉彥 劉艷 周紫章 翟一凡



摘要:斑翅果蠅(Drosophilasuzukii)是一種農業害蟲,在世界范圍內分布廣泛,可對多種水果產生不可逆損害,造成巨大經濟損失。本試驗對斑翅果蠅YolkProtein1(Ds-YP1)基因進行原核表達,并對Ds-YP1蛋白的功能結構域和信號肽進行預測,克隆多肽并構建pET-32a-c(+)-Ds-YP1原核表達載體,IPTG誘導表達并免疫小鼠制備抗體;用成功制備的抗體進行斑翅果蠅蛋白WesternBlot驗證。結果表明構建的pET-32a-c(+)-Ds-YP1表達載體在大腸桿菌中表達了His-Ds-YP1融合蛋白,通過免疫小鼠成功制備并純化了Ds-YP1多克隆抗體。制備的Ds-YP1多克隆抗體不但能識別His-Ds-YP1融合蛋白,而且能識別斑翅果蠅Ds-YP1蛋白,具有較好的特異性。本研究成功制備了斑翅果蠅Ds-YP1的多克隆抗體,對進一步研究YolkProtein蛋白作用機制具有重要意義。
關鍵詞:斑翅果蠅;卵黃蛋白;原核表達;抗體
斑翅果蠅(Drosophilasuzukii)是一種入侵性、破壞性作物害蟲,最早在日本發現[1],雄性在每個翅膀上均有一個黑點[2,3]。斑翅果蠅不像其他大多數果蠅物種只攻擊腐爛的水果[4,5],其更喜歡未受損的成熟水果[4,5];利用鋸齒狀產卵器可刺穿果實(如櫻桃和一些漿果)相對堅硬的表皮并在其中產卵,在2008年首次侵入美國時就對其造成了約50億美元的損失[6-8]。
卵黃蛋白(yolkprotein)是一種對生殖極為重要的糖脂蛋白,是所有卵生脊椎動物和無脊椎動物卵黃的主要成分[9,10]。其前體在雌性脂肪體內合成,釋放到循環系統中,并通過受體介導的內吞作用運輸到生長中的卵母細胞中,或由卵巢濾泡細胞直接合成作為營養源被利用;經20-羥基蛻皮酮調控后也可在少數雄性體內合成,但量比雌蟲低數千倍[11,12]。大多數果蠅體內存在至少兩種YP基因,有些果蠅例如黑腹果蠅(D.melanogaster)存在3種[13]。在表達方面YP1表達含量往往更高,其次是YP2,其在卵巢與脂肪體中的表達也并不一致[14]。
有關果蠅的生殖滯育已有報道,但關于斑翅果蠅的滯育卻鮮有人研究[15]。本實驗室前期已對其滯育溫度、光周期與滯育率進行研究,在此基礎上繼續對其滯育機制進行探究[16,17]。本研究通過對斑翅果蠅的YolkProtein1(Ds-YP1)基因進行分析,構建pET-32a-c(+)-Ds-YP1原核表達載體,表達并制備蛋白特異性抗體,以期為卵黃蛋白的作用機制研究提供支持。
1材料與方法
1.1供試昆蟲
斑翅果蠅采自山東省泰安市櫻桃園,由本實驗室人工飼養。飼養條件:溫度(25±1)℃,濕度(50±5)%,光周期16L∶8D。成、幼蟲皆用人工飼料飼喂,成蟲期補給20%糖水。人工飼料配方:麥麩39.1g、蔗糖20.8g、啤酒酵母5.3g、山梨酸0.17g、尼泊金乙酯0.1g、對羥基苯甲酸甲酯0.07g、抗壞血酸0.09g、維生素溶液1mL、水100mL
1.2序列分析
斑翅果蠅YP1基因序列下載自NCBI,序列AccessionNO.為XM_017081211.2。分別用InterProScan5(http://www.ebi.ac.uk/Tools/pfa/iprscan5/)和signalP4.1Server(http://www.cbs.dtu.dk/services/SignalP/)對Ds-YP1的功能結構域和信號肽進行預測。用DNAMAN(version6)進行序列翻譯及展示。
1.3引物設計
用VectorNTI軟件與NCBI的Primer-BLAST進行引物設計(表1)。由上海生工生物工程技術服務有限公司合成。
1.4RNA提取和cDNA合成
取10只斑翅果蠅置于1.5mL離心管中,放于液氮速凍研磨,按R6934-01TotalRNAKitⅡ(Omega)試劑盒使用說明提取RNA,最后加入20μLNuclease-FreeWater洗脫RNA,-80℃保存。
按TQ2501-02M-MLVFirstStrandcDNASynthesisKit(Omega)試劑盒說明書合成cDNA,合成產物保存于-20℃冰箱備用。
1.5PCR擴增
以斑翅果蠅cDNA為模板進行PCR擴增,擴增體系(50μL):2×phantaMaxBuffer25μL,dNTPMix(10mmol/Leach)1μL,上下游引物Ds-YP1-1F/R或Ds-YP1-2F/R各2μL,cDNA1μL,phantaMaxSuper-FidelityDNAPolymerase1μL,Nuclease-FreeWater補足至50μL。反應程序:94℃預變性5min;94℃變性30s,62℃退火30s,72℃延伸1min,共35個循環;72℃延伸5min。PCR結束后用1%瓊脂糖凝膠電泳檢測(120V、25min)。
1.6重組表達載體構建
擴增產物經1%瓊脂糖凝膠電泳檢測后,切取凝膠中的目標條帶,用膠回收試劑盒(北京聚合美生物科技有限公司)進行回收。將膠回收產物與pET-32a-c(+)(Novagen)一起用限制性內切酶EcoRⅠ(Thermo)、NotⅠ(Thermo)進行雙酶切。將酶切后DNA產物與載體產物膠回收后,用T4連接酶(Takara)進行連接,組成pET-32a-c(+)-Ds-YP1重組表達載體。
將酶連產物轉入EscherichiacoliDH5α(天根生化科技有限公司),在含有氨芐青霉素抗性的LB固體培養基上涂板,用無菌玻璃涂布器將菌液輕輕涂布均勻,封口膜封口后,倒置于37℃恒溫培養箱培養12~16h。挑取單菌落至LB液體培養基中,37℃過夜培養,進行陽性克隆鑒定,體系為10μL:2×TaqMasterMix5μL,T70.4μL,T7Terminator0.4μL,菌液1μL,ddH2O補足至10μL,反應程序同1.5。
將鑒定正確的菌液送上海生工生物工程技術服務有限公司進行測序,測序結果用VectorNTI軟件進行比對。選擇測序結果正確的克隆提取質粒(Vazyme,FastPurePlasmidMiniKit),-20℃保存。
1.7蛋白誘導表達
將重組質粒轉入E.coliBL21(天根生化科技有限公司)中,在含有氨芐青霉素抗性的LB固體培養基上%板,待長出菌落后挑單菌落于LB液體培養基中,37℃、220r/min培養,菌液OD600值達到0.6左右時加入IPTG誘導劑至終濃度0.1mmol/L,37℃、150r/min培養6h,之后4℃、8000r/min離心10min收集菌體。
在未誘導與誘導6h樣品菌體中加1×PBS(0.2g/mL),振蕩懸浮后用超聲波細胞粉碎機(寧波新芝生物科技有限公司)進行破碎:功率20%,超聲每次2s,間隔3s,破碎至菌體均勻清亮。
離心收集上清和沉淀,上清作為蛋白上清樣品;沉淀經含8mol/L脲的1×PBS溶解后再離心得到上清液作為包涵體;超聲破碎后未經離心的樣品作為混合物樣品。
將蛋白上清、包涵體和混合物樣品在蛋白電泳儀(Bio-Rad)上進行SDS-PAGE電泳,恒壓120V電泳至溴酚藍條帶跑出膠板下邊緣關閉電泳儀。將蛋白膠于考馬斯亮藍R250染色液中染色30min,過夜脫色后觀察蛋白表達情況。
1.8WesternBl
在SDS-PAGE電泳結束后,用濕轉電泳儀(Bio-Rad)在冰浴條件下進行轉膜,300mA恒流轉膜120min。結束后將0.45μmPVDF膜(MilliPore)浸入封閉液中,搖床常溫孵育1h,用TBST漂洗10min;加入用BSA稀釋10000倍的MouseantiHis-TagmAb(ABclonal)4℃過夜孵育;回收一抗后再用TBST漂洗3遍,每遍10min;加入用BSA稀釋10000倍的辣根酶標記山羊抗小鼠IgG(北京中杉金橋生物技術有限公司),常溫搖床孵育2h;TBST漂洗4遍,每遍10min;最后使用極超敏化學發光試劑盒(山東思科捷生物技術有限公司)顯色,于ChemiScope系列熒光及化學發光成像系統(上海勤翔科學儀器有限公司)中顯影。
1.9抗體制備
目的蛋白條帶切膠-20℃保存。將蛋白膠條放入干凈研缽中,研磨至黏稠狀,加0.9%生理鹽水稀釋備用。SPF級KM小鼠(濟南朋悅實驗動物繁育有限公司)7只,每只20g左右,喂養3天后用1mL針管45度角注射腹部位,每次每只小鼠注射300~500μL,注意排盡針管空氣。每周免疫一次,連續4周,第5周摘眼球取血。
血液放于1.5mL離心管中,立即放入37℃水浴鍋1h,然后4℃放置6h,之后4℃、12000r/min離心15min,取上清,液氮速凍后放于-80℃凍存。
1.10果蠅總蛋白提取
提取蛋白全程均在冰上操作,研磨棒需預冷。取15只斑翅果蠅放入液氮速凍,加200μL研磨液進行冰浴研磨,然后冰上靜置30min,每隔幾分鐘混勻一次;離心機預冷至4℃,12000r/min離心5min,取上清;加入蛋白上樣緩沖液100℃金屬浴5min;-20℃保存備用。
2結果與分析
2.1序列分析及克隆
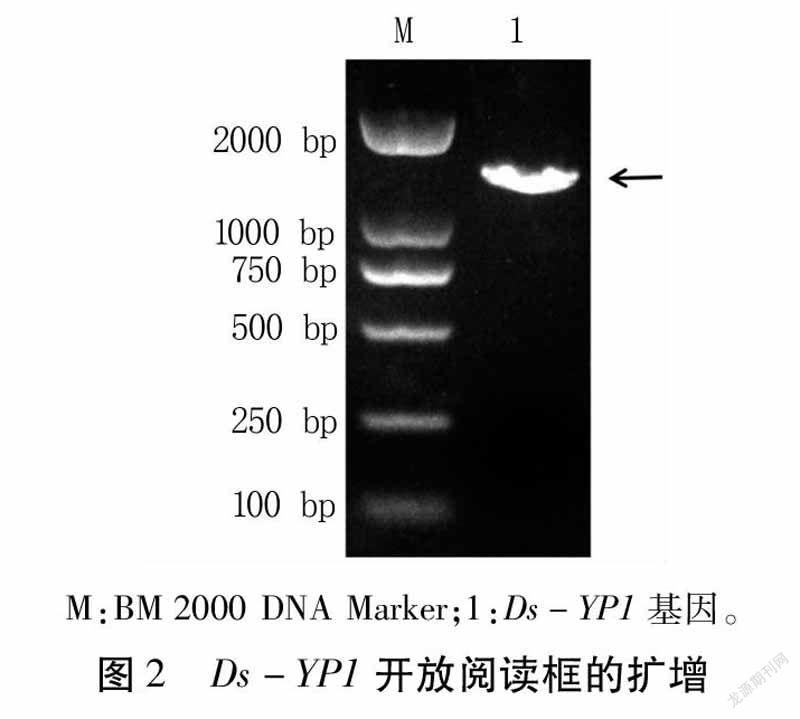
Ds-YP1序列開放閱讀框為1320bp,編碼蛋白共439個氨基酸。Ds-YP1蛋白具有信號肽(1~19aa)和完整的vitellgenin功能結構域(129~410aa,圖1),去除信號肽后經軟件預測分析,分子量為46.96kD。對完整的開放閱讀框進行PCR克隆及測序,結果表明,序列長度與數據庫序列一致,且無突變堿基(圖2)。
2.2重組表達載體的構建
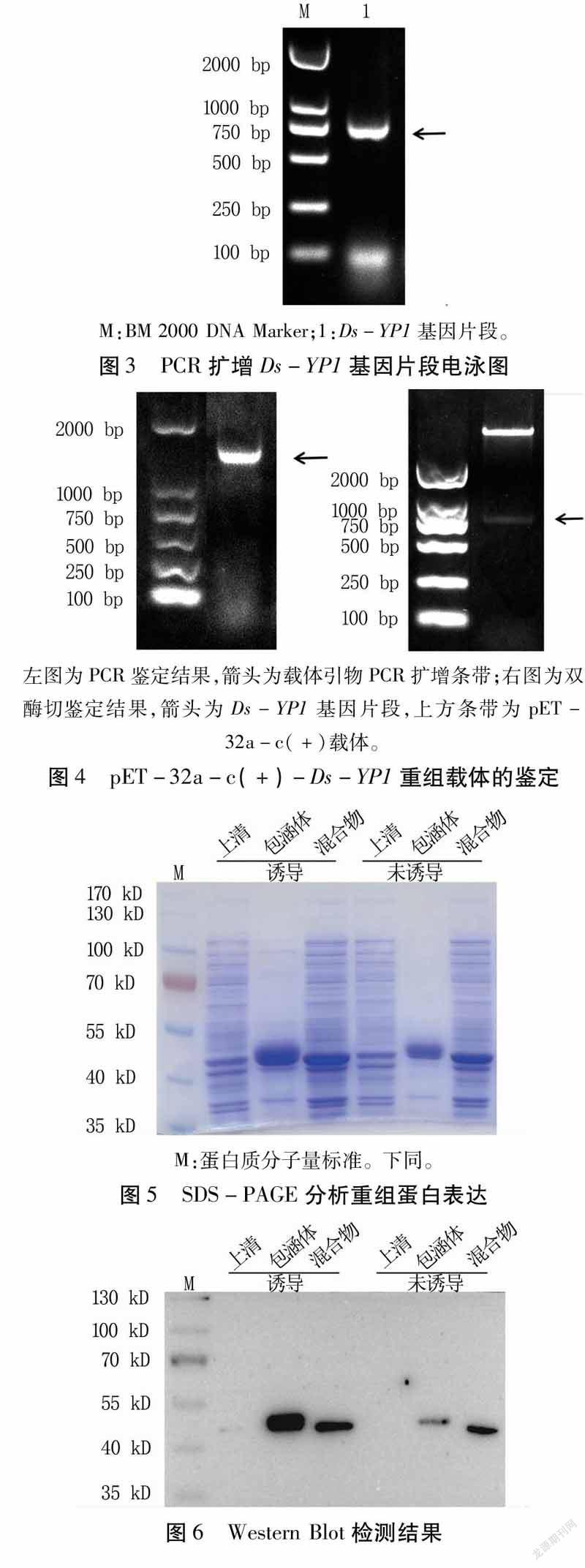
首先選取不含信號肽的部分序列進行擴增(圖3),將擴增目的片段與pET-32a-c(+)載體經EcoRⅠ、NotⅠ酶切,經連接轉化后構成重組表達載體pET-32a-c(+)-Ds-YP1。構建的載體用通用引物經PCR和雙酶切鑒定,得到的片段大小與預期一致(圖4)。測序結果發現重組表達載體中的插入片段與Ds-YP1基因的序列完全一致,且無移碼現象。以上結果說明,pET-32a-c(+)-Ds-YP1重組表達載體構建成功,可以用來誘導表達重組蛋白。
2.3重組蛋白在大腸桿菌中誘導表達
將未誘導與誘導樣品的蛋白上清、包涵體和混合物樣品分別進行SDS-PAGE電泳,考馬斯亮藍R250染色后,發現目的蛋白在蛋白上清、包涵體及混合物中都有表達,且包涵體中表達量最高(圖5)。從WesternBlot檢測結果也可看出,目的蛋白與His抗體發生特異結合且與預期大小一致,證明重組蛋白正確表達(圖6)。
2.4抗體制備與檢測
將包涵體中的重組蛋白經SDS-PAGE電泳后切膠回收免疫KM小鼠,4周后收集血清,檢測血清中的抗體Anti-Ds-YP1。將血清稀釋5000倍后,檢測未誘導與誘導樣品,結果顯示,條帶單一且效價高(圖7)。隨后用制備的Anti-Ds-YP1抗體WesternBlot檢測斑翅果蠅蛋白樣品,結果顯示,在抗體稀釋18000倍時,條帶清晰,信號強(圖8)。
以上結果表明制備的Anti-Ds-YP1特異性強且效價高,結果與預期相符,可用于后續試驗。
3討論與結論
卵黃蛋白是卵黃發生時沉積在卵內供胚胎發育營養所需的卵內貯藏蛋白。昆蟲的卵黃蛋白主要分為卵黃蛋白原(vitellogenin)、卵黃多肽(yolkpolypeptides)和小卵黃蛋白(minorYP)三種[18]。其中卵黃蛋白原是一類大分子量的糖脂復合蛋白,與非昆蟲的卵黃蛋白原進化上同源,可能起源于共同的祖先[19]。卵黃多肽存在于高等雙翅目昆蟲中,功能等同于其它昆蟲卵黃蛋白原,但是分子量較小,約為45kD[20]。小卵黃蛋白是在鱗翅目昆蟲卵內分布的一類特異性蛋白[21]。本研究中,斑翅果蠅的卵黃蛋白大小為46.96kD,屬于卵黃多肽。
雖然卵黃多肽功能上與卵黃蛋白原相似,但其進化上卻完全不同,卵黃蛋白原與脊椎動物的血清載脂蛋白B(apolipoproteinB)具有較高的同源性,卵黃多肽與肝酯酶(hepaticlipases)和胰酯酶(pancreaticlipases)進化上同源。在黑腹果蠅中有3種YP,主要在脂肪體和卵巢泡細胞中合成,但在不同組織中的分布和表達不同[22]。果蠅(Drosophilagrimshawi)中也有3種YP,在脂肪體中YP1合成最多,而YP2和YP3較少;在卵巢內,YP2合成最多,YP1較少,而YP3幾乎不存在[23]。本試驗用抗體檢測斑翅果蠅YP(圖8),發現在目的條帶上方存在條帶,這可能是斑翅果蠅的其它YP,其不同YP的含量不同可能與果蠅的發育時期和組織分布有關。
本試驗通過構建重組質粒,轉化大腸桿菌后進行原核表達,并用小鼠進行抗原免疫,成功獲得了高質量抗體。后續研究將利用該抗體對卵黃蛋白在斑翅果蠅中的功能進行分析。本研究結果將有助于對斑翅果蠅生殖發育等機制開展更深入的研究,為果蠅防治工作提供新的途徑。
