PEComa臨床病理分析與影像診斷
2022-05-10 05:43:50張泳欣肖學紅唐秉航黎衛黃紅星袁潤強盧揚柏
國際醫藥衛生導報 2022年10期
張泳欣 肖學紅 唐秉航 黎衛 黃紅星 袁潤強 盧揚柏
1中山市人民醫院影像中心,中山 528403;2中山市人民醫院泌尿外科,中山 528403
血管周上皮樣細胞腫瘤(perivascular epithelioid cell tumor,PEComa)屬于較為罕見的間葉源性腫瘤,由具有特殊的組織學和免疫組織化學特征的血管周圍上皮樣細胞組成。2002年,世界衛生組織(WHO)將PEComa定義為“由組織學和免疫組織化學上獨特的血管周上皮樣細胞組成的間葉性腫瘤[1]。PEComa可分為良性、惡性、惡性潛能3類,以良性居多,好發于女性[2]。PEComa可發生于腎、肝、肺、子宮、卵巢、胃腸道、胰腺、前列腺和軟組織等部位。據文獻查閱,涉及腎臟惡性腫瘤的PEComa研究仍然很少[3-5]。筆者收集2010—2021年中山市人民醫院確診的1例腎臟惡性PEComa,結合2000—2021年期間中國知網(CNKI)、萬方數據庫報道的文獻探討其臨床表現、病理及免疫表型特點、診斷與鑒別診斷、治療及預后,旨在提高對本病的認識。
資料與方法
1、臨床資料
回顧性分析2021年12月30日中山市人民醫院收治的1例腎臟惡性PEComa患者的臨床病理特點,經病理科最終確診,符合WHO對PEComa的診斷分類。在CNKI、萬方數據庫搜索2000—2021年期間,關鍵詞:“腎臟”“上皮樣血管周細胞腫瘤”“血管周上皮樣細胞腫瘤”“PEComa”文獻,總共得到18篇文獻,239例患者,明確指出惡性的有20例,所有病例均進行不定期隨訪。
2、方法
2.1、掃描方法 采用Siemens Somatom 256排螺旋CT掃描儀(德國),囑患者仰臥,掃描范圍自膈頂至雙側髂前上棘水平。行平掃及增強掃描,掃描參數如下:管電壓120 kV,管電流200~300 mA,層厚5 mm,層間距5 mm,重建層厚1 mm,重建矩陣256×256。增強掃描:經肘正中靜脈注射對比劑1.5 ml/kg體質量,流率3.0~3.5 ml/s。6例在注射對比劑30 s、60 s、300 s時行皮髓質期、實質期及排泄期增強掃描。將原始數據傳輸至CT機配套后處理工作站進行處理。
2.2、病理檢查方法 采用4%中性甲醛液固定,常規脫水,經石蠟包埋后切割成4~5μm切片,經蘇木精-伊紅染色(HE染色)后用光鏡觀察。免疫組織化學染色采用En Vision兩步法,具體步驟參照說明書進行。所用抗體黑色素瘤相關抗原(HMB45)、T細胞識別的黑色素瘤抗原(Melan-A)、酸性鈣結合蛋白(S100)、鋅依賴性細胞膜金屬蛋白酶(CD10)、Ki67等及En Vision試劑盒均購自福州邁新生物技術開發有限公司。
3、圖像分析
由2位經驗豐富的影像診斷醫師(1名主治醫師和1名副主任醫師)進行閱片、分析及記錄,并與相關專科醫生討論達成一致。影像學評價包括腫瘤的位置、大小、形態、密度、強化特點、有無鈣化及液化壞死、腫瘤邊緣、是否有子灶、是否轉移及其與周圍組織關系。
結 果
1、臨床資料特點
本例患者為女性,58歲,在2021年11月無明顯誘因出現腹脹而檢查發現病變。回顧文獻資料,一共239例患者,男54例(22.6%)、女185例(77.4%),其中20例明確指出惡性;臨床表現無特異性,常見的臨床表現為腰痛、腰脹;其次是體檢偶爾發現;再者較少見的就診原因為腫瘤破裂出血急診就診、血尿、腹部包塊、其他地方病變檢查無意中發現等。本例患者位于左腎,經腹腔鏡病灶根治術切除,術后病灶經病理證實為惡性PEComa,本研究病例在隨訪期內,未見轉移或復發。
2、影像學表現
腎臟惡性PEComa的影像學表現具體見圖1。
3、病理特點分析
鏡檢:腫瘤細胞呈泡巢狀、乳頭狀增生,腫瘤中見由小的薄壁血管構成網狀結構,腫瘤中央可見大片壞死。瘤細胞多角形,胞漿嗜酸,部分透明。細胞核圓形或卵圓形,染色質增多,核仁明顯,周圍纖維組織增生,纖維化、玻璃樣變及鈣化。免疫組化:Ki67 10%+、細胞角蛋白(CK)-、高分子量細胞角蛋白CK(HMW)-、P63-、CD10散在+、堿性細胞角蛋白(CK7)-、Ⅲ型受體酪氨酸激酶(CD117)-、a-甲基酰基輔 酶A(AMACR)-、轉錄因子E3(TFE3)-、波 形 蛋白(Vimentin)+、HMB45+,Melan-A+,S100-,細胞周期蛋白依賴激酶4(CDK4)-,鼠雙微體基因2(MDM2)-,突觸素(SYN)-,嗜鉻素A(CgA)-。具體見圖1G。
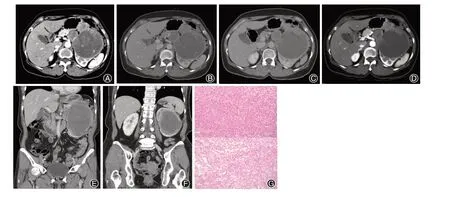
圖1 腎臟惡性血管周上皮樣細胞腫瘤患者的影像學表現及病理特點分析。A~F為左側腹膜后見分葉狀巨大腫塊,密度混雜,內側成分房狀改變,可見實性成分及斑點狀鈣化灶,實性成分輕度強化,病灶最大截面約98 mm×96 mm,與左腎及胰尾分界欠清,其余周圍結構受壓改變,腹膜后未見明顯腫大淋巴結未見腹水征象;G為病理切片(HE染色 ×100)
討 論
1、發病機制
PEComa是罕見的間葉性腫瘤,1996年Zamboni等[6]首次將其報道并命名。許多人努力研究這些腫瘤在多潛能原始細胞中的起源,特別是那些位于血管周圍區域的細胞。最近,人們提出了幾個假設[7]。一種理論認為,能夠表達黑素細胞和平滑肌表型的神經嵴未分化細胞代表PEComa細胞的起源。其次,有人提出PEComa可能來源于周胞質元素。此外,有研究認為PEComa起源于成肌細胞和平滑肌,并具有獲得性黑素細胞標志物表達[8]。
2、病理特點
組成PEComa的上皮樣細胞呈放射狀排列分布于血管周圍,胞漿透明至嗜酸性,常表達黑素細胞和肌源性標記物[9]。由于PEComa的罕見性,直到2010年才制定出區分良性和惡性腫瘤的診斷標準[10]。Folpe等[11]提出了目前被接受的良性、惡性潛能或惡性PEComa的診斷標準:良性(直徑<5 cm、低核級別、細胞密度低、有絲分裂率<1/50高倍視野、未見浸潤/壞死/血管侵犯);以下標準出現其中一個考慮惡性潛能:核異型性、多核巨細胞以及大小≥5 cm;顯示以下2種或多種情況的腫瘤被認為是惡性的:直徑>5 cm、浸潤性生長、細胞密度大、核大和著色過深、有絲分裂活性高、有絲分裂像不典型、合并凝固性壞死/血管侵犯等。
PEComa表達一種肌黑素細胞表型,其對黑素細胞和平滑肌標志物具有免疫反應性。已經證實HMB45和某些其他黑素細胞標記物是PEComa的免疫組織化特點[11-15];與其最相關的是小眼癥轉錄因子(MTF)、Melan-A/Mart-1和HMSA-1[16]。大約70%的病例出現了平滑肌肌動蛋白(SMA)+、波形蛋白和/或結締組織蛋白+、組織蛋白酶K+等[17-20]。Fadare等[21]在近期的文獻綜述中發現,在子宮出現的PEComa中表達的免疫組化指標及陽性百分比是HMB45 100%+,SMA 73%+,波形蛋白56%+,CD10 25%+,Melan-A 24%+,CD117 9%+,高度糖基化Ⅰ型跨膜蛋白(CD34)5%+,S100 3%+,角蛋白3%+等。其中,HMB45+為特征性表現。本研究病例及搜索到的文獻HMB45均為陽性。
3、臨床特征
PEComa好發于女性[21]。2004年WHO將其歸為具有惡性的潛能[22]。有研究認為PEComa有1/3的病例會出現局部復發和/或遠處轉移[22]。惡性者的常見起源部位包括腎臟和腹膜后軟組織,以及女性生殖道的子宮和宮頸[23]。惡性PEComa伴隨著復發和/或轉移,轉移部位最常見的是肝臟[24-25]。
4、影像學表現
影像學表現缺乏特異性,CT、磁共振成像(MRI)、超聲檢查(US)和正電子發射型計算機斷層顯像(PET-CT)等影像學研究可以幫助檢測腫瘤的位置、大小以及與周圍組織的關系。術前影像學檢查大多表現為混雜密度,邊界相對清楚的病灶,增強后表現為明顯強化,因此容易誤診為惡性腫瘤,在無鄰近器官侵犯或局部轉移的情況下,較難與良性者相鑒別,惡性者最常見的CT表現是內臟或腹膜后腫塊邊界清楚,與同層面肌肉相比,平掃為低/等密度,明顯強化,需要注意的是,強化模式以不均勻強化更常見[24]。點狀鈣化相對少見。在MRI上,相對于同層面骨骼肌,大多數腫瘤T1WI表現為等/稍低信號。有研究提示,若病灶具有T2WI低信號,彌散加權成像(DWI)受限擴散,明顯強化(快進快出)等特點,需考慮PEComa[26-27]。本例患者為位于左側腹膜后的巨大分葉狀腫塊,密度混雜,內部見分房,可見實性成分(輕度強化)及斑點狀鈣化灶。林樂等[28]報道的10例腎血管周上皮樣細胞腫瘤,大部分病例表現為B超高回聲,CT下密度混雜[29-30]。
5、診斷
在病理學和影像學檢查中,此類腫瘤很容易被誤診為腎細胞癌[31-32]。在圖像上未發現脂肪的PEComa將難以與乏脂肪的血管平滑肌脂肪瘤的其他亞型以及腎細胞癌相鑒別[33]。腎細胞癌中的脂肪成分通常不明顯,若合并鈣化,則通常較大且粗糙。含有脂肪但無鈣化的腎細胞癌極為罕見[34-35]。
影像學的某些表現對提示患者預后有一定價值,包括腫瘤大小、是否存在其他血管平滑肌脂肪瘤、是否有轉移、淋巴結腫大、腎外侵犯、腎盂受累和腎靜脈受累等[36]。Cui等[37]報告1例巨大腎PEComa伴巨大腹膜后轉移性淋巴結病。在Froemming等[38]報道的9個病例中,術前CT和MRI顯示腫大的淋巴結,其中1個被病理證明是轉移性PEComa;另外5個病例中出現了向腎竇的延伸,這與腎細胞癌吞噬腎竇脂肪相似。
6、治療
手術是PEComa的主要和最有效的治療方法。惡性者除手術切除外,化療和放療效果仍不明確,預后差[39]。本病采用腹腔鏡腎癌根治術(左側)+胰腺部分切除+脾臟切除術+降結腸部分切除術+橫結腸端側吻合+腹膜后淋巴結清掃術+腸粘連松解術,在術后的隨訪期間內,暫無出現復發/轉移,但無病生存期的長短仍需繼續隨訪。
綜上所述,原發性腎臟惡性PEComa是罕見的間葉源性腫瘤,組織學和免疫組化有一定特征,影像學檢查具有一定的特點。鑒于惡性PEComa的發病機制和生物學行為目前尚不清楚,因此手術仍然是最佳治療的主要方法。
利益沖突 所有作者均聲明不存在利益沖突
