木論喀斯特常綠落葉闊葉混交林土壤真菌群落多樣性特征
2022-05-14 03:13:10谷俊錕曾馥平宋同清彭晚霞蘇樑杜虎
生態科學 2022年3期
關鍵詞:分類
谷俊錕, 曾馥平 宋同清 彭晚霞 蘇樑 杜虎*
木論喀斯特常綠落葉闊葉混交林土壤真菌群落多樣性特征
谷俊錕1,2,3, 曾馥平1,2, 宋同清1,2, 彭晚霞1,2, 蘇樑1,2, 杜虎1,2,*
1. 中國科學院亞熱帶農業生態研究所亞熱帶農業生態過程重點實驗室, 湖南長沙 410125 2. 中國科學院環江喀斯特生態系統觀測研究站/廣西喀斯特生態過程與服務重點實驗室, 廣西環江 547100 3. 湖南農業大學資源環境學院, 湖南長沙 410125
土壤微生物在森林生態系統中起著重要作用, 為了解喀斯特常綠落葉闊葉混交林土壤真菌多樣性, 采用Illumina Hiseq高通量測序技術對木論喀斯特森林土壤真菌群落多樣性進行了初步研究。研究結果表明: 在82個采樣點內, 隨著采樣點增加, 檢測出不同分類水平的土壤真菌類群逐步增多; 土壤樣點數達到82個時, 檢測出的土壤真菌類群達6門32綱126目336科886屬和132616個種(OTU); 82個土壤樣品中所檢測出的真菌類群平均有5.41門22.13綱71.09目144.89科216.66屬和1256.44個OTU, 其中門、綱、目分類水平上的優勢類群(所占比例)分別為子囊菌門()(50.09%)、接合菌綱()(34.54%)、被孢霉目()(22.65%)。取樣數量對所檢測土壤真菌類群數據有顯著影響, 在門、綱、目、科的分類水平上, 1個、15個、48個、73個樣品基本上可以代表82個樣品檢測出來不同分類水平的真菌多樣性; 在屬和OTU的分類水平上, 當樣品數達到82個時檢測出來的響應類群數仍在不斷增加。該研究為進一步了解喀斯特森林土壤真菌多樣性與植物多樣性和生境的關系提供了基礎。
土壤真菌多樣性; 高通量測序; 采樣數; 森林; 喀斯特生態系統
0 前言
微生物是土壤最活躍的組成, 在土壤中儲存了數量龐大微生物資源[1, 2]。土壤微生物參與了土壤發生、發展、發育的全過程, 在物質循環、能量轉換以及污染物降解等過程中都發揮著重要作用[3]。真菌作為土壤微生物中的重要組成部分, 可有效降解土壤中復雜組分和凋落物, 驅動養分循環和能量流動, 還可反映土壤病理過程, 是陸地生態系統的重要健康指標[4, 5], 并且和植物、細菌之間維持著重要的共生關系[6]。大量研究發現, 森林生態系統具有極其豐富的微生物資源, 且由于微生物結構相對簡單, 能對環境變化做出快速而靈敏的反應。相對于土壤細菌, 雖然森林土壤真菌的研究受到的關注偏少, 但真菌在森林生態系統中對于促進宿主植物對礦物質吸收、穩固和改善土壤結構的作用不容忽視[7-9]。在亞熱帶不同植被的影響下, 隨著生態環境改善、植被的恢復, 土壤真菌群落多樣性逐漸增加[10]。中溫帶氣候下不同林型的土壤真菌群落結構存在較大差異。在寒溫帶氣候下, 森林植被類型、土壤環境因子和林下植被對土壤真菌群落結構影響顯著[11, 12]。不同植被類型凋落物通過改變土壤理化性質進而影響土壤真菌多樣性[13]。充分了解森林生態系統土壤真菌群落多樣性對認識生態系統結構、功能與過程, 評價生態系統服務等具有重要的意義。在熱帶和溫帶, 經大量研究發現, 土壤真菌門類群含量一致豐富的是子囊菌門[14-16], 土壤真菌屬類群豐富的是被孢霉屬[17]。據估算, 全球范圍內真菌種數可達80-510萬, 而目前已知大約有10萬種[5]。因此更加全面的反映微生物信息顯得尤為重要。隨著分子生物學技術的發展, 從早先的平板培養法發展到高通量測序技術的應用, 使得更為簡單、快速、準確的獲取土壤微生物信息成為可能[2]。獲得一定數量且具有代表性的森林土壤樣品是森林土壤微生物群落調查的首要工作。在目前的研究當中對每個樣地或樣方內需要采集多少數量的樣品并沒有統一標準[18]。在考慮代表性和可操作性前提下, 究竟多少個土壤樣品才能充分反映森林土壤微生物群落信息?這是一個基礎但重要問題。
位于我國西南喀斯特地區的廣西木論國家級自然保護區與毗鄰的貴州茂蘭國家級自然保護區連片保存著目前世界上連片面積最大、保存最完好、原生性最強的喀斯特非地帶性植被——喀斯特常綠落葉闊葉混交林。該區域也是我國生物多樣性3個特有分布中心之一, 且位于我國植物區系相匯交錯區和交接過渡帶, 其植被和生境呈現高度的異質性[19, 20], 是進行喀斯特地質背景下生物多樣性相關研究的理想場所。本研究基于Illumina HiSeq測序平臺對位于木論國家級自然保護區內25 ha大樣地土壤真菌多樣性進行了初步研究, 分析了土壤真菌門、綱、目、科、屬等不同分類水平優勢類群和各類群相對豐度, 并探討了取樣數量對土壤真菌多樣性檢測結果的影響, 以及在該生境內多少個土壤樣品可以代表不同分類水平的真菌多樣性, 其結果將有助于進一步回答土壤真菌多樣性及其與植物多樣性的關系。
1 材料與方法
1.1 研究區概況
木論國家級自然保護區位于廣西環江毛南族自治縣西北部, 地理坐標為107°54′01—108°05′51 E, 25°07′01—25°12′22 N之間, 屬于中亞熱帶石灰巖區常綠落葉闊葉混交林生態系統, 是我國生物區系相匯交錯區和交接過渡的中心, 生境異質性極高, 區系成分復雜, 生物種類豐富, 生態環境脆弱, 是目前世界上喀斯特地貌區幸存連片面積最大、保存最完好、原生性最強的喀斯特森林。該區域為中亞熱帶季風氣候, 年平均氣溫19.3℃, 年均降水量1529 mm。區內土壤主要為非地帶性石灰土。2014年參照CTFS(Center for Tropical Forest Sciences)森林樣地建設技術要求在該保護區內建成了25 ha (500 m×500 m)森林動態監測樣地。
1.2 土壤樣品采集
監測樣地按網格法劃分成了625個20 m×20 m樣方, 2016年秋季在監測樣地中相對均勻地選取了82個樣方進行土壤樣品采集(圖1), 在各樣方中心點附近用土鉆采集5個0—10 cm表層土壤混合成一個樣品, 樣品去除根系、石頭等雜質后, -80 ℃保存用于DNA提取和后續實驗分析。
1.3 DNA提取、擴增和高通量測序
土壤微生物宏基因組DNA提取采用的專用試劑盒(FastDNA?SPIN Kit for Soil, MP)。采用引物ITS5-1737F(GGAAGTAAAAGTCGTAACAAGG), ITS2-2043R(GCTGCGTTCTTCAT CG A TGC)對真菌ITS基因的ITS1區進行PCR擴增。PCR反應體系30 μL: 15 μL Phusion Master Mix(2×), 3 μL(6μM) Primer(2μM), 10 μL(5—10 ng)gDNA(1 ng/μL), 2 μL H2O[21]。PCR反應條件為: 98℃預變性1 min; 30個循環包括(98 ℃, 10 s; 50 ℃, 30 s; 72 ℃, 30 s); 72 ℃, 5 min。依據PCR產物濃度, 將所有擴增成功的PCR產物等量混合, 再經定量等質量控制后, 將質量合格的PCR產物進行DNA文庫的構建, 采用Illumina Hiseq平臺進行雙末端250 bp測序(委托諾禾致源生物信息科技有限公司完成)。
1.4 高通量測序數據處理與統計分析
根據Barcode序列和PCR擴增引物序列從下機數據中拆分出各樣品數據, 截去Barcode和引物序列后使用FLASH(V1.2.7, http://ccb.jhu.edu/software/ FLASH/)[22]對每個樣品的reads進行拼接, 過濾掉低質量的序列。使用Qiime(V1.7.0, http://qiime.org/ scripts/split_libraries_fastq.html)[23]的Tags質量控制流程做聚類及多樣性分析。其中, 利用Uparse軟件(Uparse v7.0.1001, http://drive5.com/uparse/)[24]對所有樣品進行聚類, 依據慣例以97%的一致性將序列聚類成為OTUs[25], 依據其算法原則, 篩選出OTUs中出現頻數最高的序列作為OTUs的代表序列, 用RDP對其進行系統分類[26]。再用QIIME進行單樣品組成分析[23], 得到樣品在不同分類水平上的真類群組成及相對豐度的數據, 將各分類水平: Phylum(門)、Class(綱)、Order(目)、Family(科)、Genus(屬)上最大豐度排名前10的物種, 生成物種相對豐度圖[27]。

圖1 木論森林動態監測樣地土壤真菌采樣點位置
Figure 1 Location of soil fungi sampling points in Mulun Forest Dynamic Monitoring Plot
在上述基礎上制作采樣點數–土壤真菌類群數曲線。具體的做法是: 從82個樣品中隨機抽取1個樣品, 記下這個樣品中包含的真菌類群數目; 然后從剩下的81個樣品中再隨機抽取1個樣品數據, 和第一個抽到的樣品數據合并, 記下這2個樣品中包含的真菌類群數目; 依此類推, 直至采樣點的數目達到最大; 重復99次取平均值, 制作采樣點數–土壤真菌類群數曲線。
本研究其他數據分析與圖形制作使用Origin 2016和R3.5.1軟件(R-development Core Team, 2018)完成。
2 結果與分析
2.1 土壤真菌分類
對木論樣地的82個土壤樣品進行高通量測序, 共產生132616個OTU。與數據庫比對不上的OTU即未分配的部分(Unassigned)約占總數的4.414%。82個土壤樣品中平均有1256.44 ±493.32個OUT, 其中單個樣品最多為3359個、最少為653個, 分屬于6門32綱126目336科886屬, 真菌分類群詳細信息見附錄1(不包含未分配的部分; 各個分類水平上未有明確分類名稱信息的也作為一個計入在內)。
82個土壤樣品中平均有真菌5.41 ±0.51門, 其中單個樣品最多的有6門, 最少的有5門(圖2)。相對豐度最高的5個門所占比例總和達到了99.56%, 分別是子囊菌門(Ascomycota)(50.09%)、接合菌門(Zygomycota)(42.13%)、擔子菌門(Basidiomycota) (6.86%)、壺菌門(Chytridiomycota)(6.01%)和球囊菌門(Glomeromycota)(0.26%)(圖3)。
82個土壤樣品中平均有真菌22.13 ±2.43綱, 其中單個樣品最多的有26綱, 最少的有18綱(圖2)。相對豐度最高的5個綱所占比例總和為71.97%, 分別是接合菌綱(Zygomycota)(34.54%)、座囊菌綱(Dothideomycetes) (15.51%)、糞殼菌綱(Sordario-mycetes)(14.04%)、傘菌綱(Agaricomycetes)(4.84%)和散囊菌綱(Eurotiomycetes)(3.05%)(圖3)。
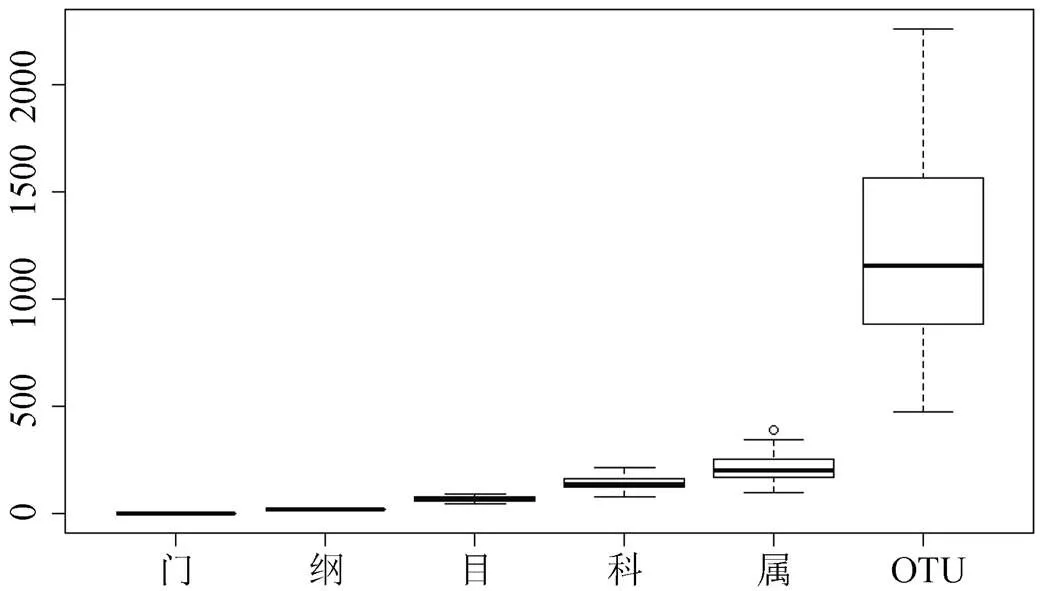
圖2 不同分類水平上真菌數量
Figure 2 The quantity of soil fungal at different taxonomical levels
82個土壤樣品中平均有真菌70.09±10.82目, 其中單個樣品最多的有94目, 最少的有48目(圖2)。相對豐度最高的5個目所占比例總和為39.42%, 分別是被孢霉目(Mortierellales)(22.65%)、肉座菌目(Hypocreales)(7.31%)、格孢菌目(Pleosporales) (4.21%)、炭角菌目(Xylariales)(2.71%)和傘菌目(Agaricales)(2.56%)(圖3)。
82個土壤樣品中平均有真菌144.89±30.02科, 其中單個樣品最多的有219科, 最少的有83科(圖2)。相對豐度最高的5個科所占比例總和為14.59%, 分別是被孢霉科(Mortierellaceae)(5.07%)、格孢菌科(Incertae_sedis_Pleosporales)(3.16%)、發菌科(Tri-chocomaceae)(2.22%)、炭角菌科(Incertae_sedis_ Xylariales)(2.09%)和古生菌科(Archaeorhizomy-cetaceae)(2.05%)(圖3)。
82個土壤樣品中平均有真菌216.66±65.73屬, 其中單個樣品最多的有392屬, 最少的有99屬(圖2)。相對豐度最高的5個屬所占比例總和為9.28%, 分別是被孢霉屬(Mortierella)(3.68%)、小畫線殼屬(Monographella)(1.93%)、白粉菌屬Microidium (1.23%)、周刺座霉屬(Volutella)(1.23%)和青霉屬(Penicillium)(1.21%)(圖3)。
2.2 取樣數對土壤真菌多樣性的影響
在各個分類水平上, 隨著取樣點的增加, 檢測出的土壤真菌類群的數目也在不斷增加。當取樣點達到82個時, 檢測出的土壤真菌類群的數目為最大, 分別為6門32綱126目336科886屬132616個OUT(圖4)。
在門的分類水平上, 當取樣點數為1個時, 約能檢測到5個門; 取樣點數在2—82區間時, 門數僅增加1個達到6個。
在綱的分類水平上, 取樣點數在1—8區間時, 檢測到的真菌綱數量隨著取樣點數量增加而快速增加; 取樣點數超過8后, 綱的數量增速減慢; 當取樣點數為15個時, 約能檢測到31個綱; 取樣點數在16—82區間時, 綱數僅增加1個達到32個。
在目的分類水平上, 取樣點數在1—25區間時, 檢測到的真菌目數量隨著取樣點數量增加而快速增加; 取樣點數超過25后, 目的數量增速減慢; 當取樣點數為48個時, 約能檢測到125個綱; 取樣點數在49—82區間時, 目數僅增加1個達到126。
在科的分類水平上, 取樣點數在1—52區間時, 檢測到的真菌科數量隨著取樣點數量增加而快速增加;
取樣點數超過52后, 科的數量增速減慢; 當取樣點數為73個時, 約能檢測到335個目; 取樣點數在74—82區間時, 科數僅增加1個達到336。
在屬的分類水平上, 取樣點數在1—70區間時, 檢測到的真菌屬數量隨著取樣點數量增加而快速增加; 取樣點數超過70后, 屬的數量增速減慢, 但增速仍較大, 且屬的增速大于門、綱、目、科的增速。
在OTU的分類水平上, 當取樣點數在1—82區間時, 檢測到的真菌OTU數量隨著取樣點數量增加一直呈現出較快增加的趨勢(圖4)。
3 討論
采用Illumina公司Hiseq測序儀對地處中亞熱帶季風氣候區的木論喀斯特常綠落葉闊葉混交林土壤真菌多樣性進行初步研究, 共產生132616個OUT, 分屬于6門32綱126目336科886屬。研究結果列出了木論樣地土壤真菌各個不同分類水平上的優勢類群及其相對豐度。其中,是最豐富的真菌類群, O'Brien 等[28]研究表明森林土壤 ITS 文庫中的和序列比例(46%和41%)相差不大, 而和占ITS克隆文庫序列的1.5%和0.5%。但本研究中序列僅占6.86%, 而序列占了 42.13%。這與 O'Brien的研究結果有一定的差異, 而Lienhard 等[29]在老撾熱帶草地中對真菌群落進行研究發現, 草地中真菌群落在門水平上的比例較高, 這與本研究結果一致。這可能由于和都屬于腐生營養土壤真菌, 在土壤真菌群落中占主導地位。但在其他有關森林土壤中真菌群落的研究中發現,在真菌群落中的比例較高, 這可能是由于土壤質地和植被類型產生的[30]。
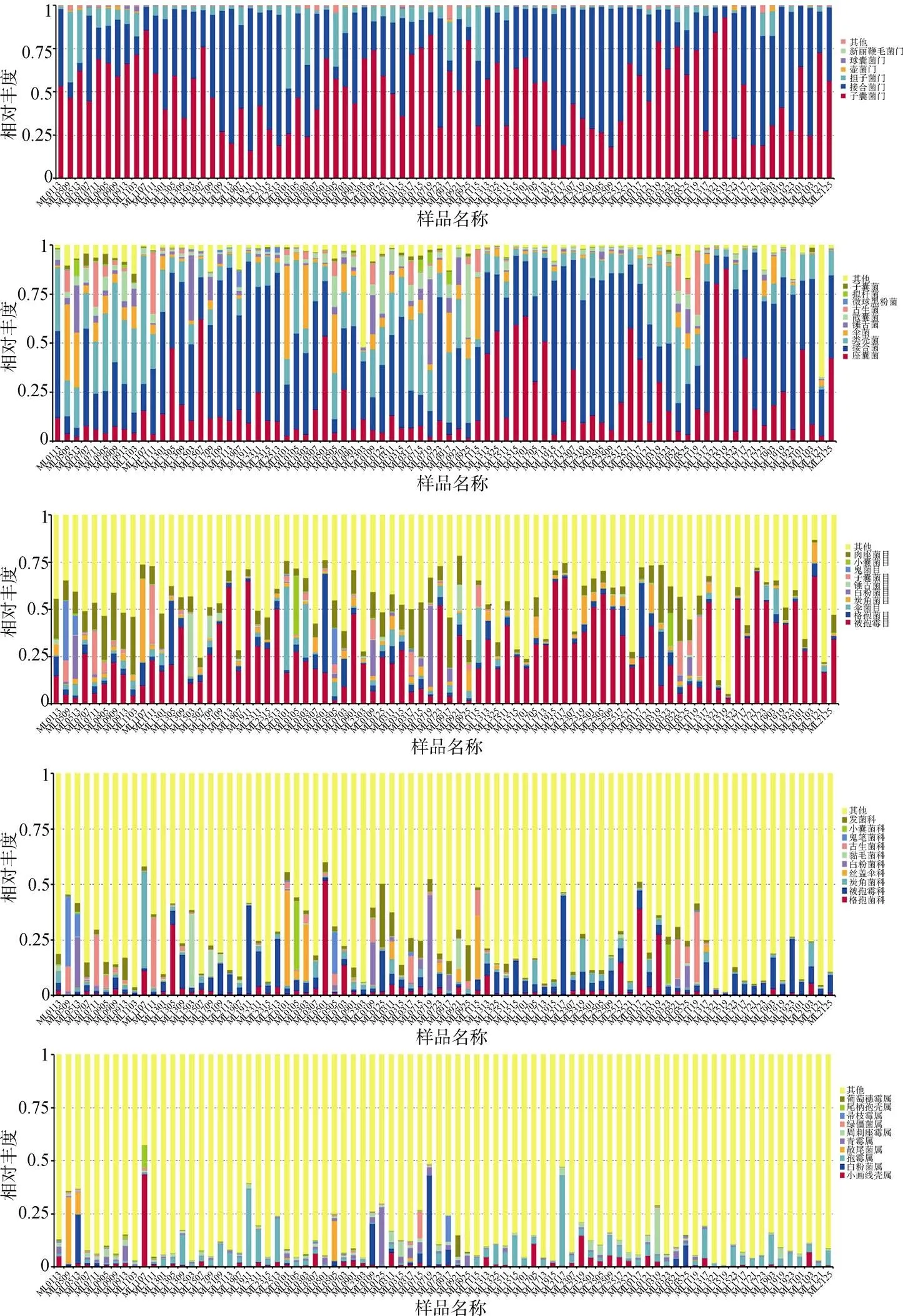
圖3 木論大樣地土壤樣品在不同水平上真菌優勢類群及其相對豐度
Figure 3 Dominant groups of fungi and their relative abundance at different levels in soil samples from the Mulun large plot.

圖4 不同分類水平上真菌類群數隨采樣點數增加的稀疏曲線
Figure 4 Rarefaction curves of the observed fungi quantity at different taxonomic levels varying with number of samples in the study area
在某種植被類型、生境類型或是某個生態系統, 所需多少土壤樣品才能較好地反映其微生物多樣性水平, 依然是一個值得探究的問題。但在采樣數量一定時, 研究人員大多還是將數個取樣點土樣混合后成一個樣品可增加樣品的代表性[31,32]。本研究中, 在不同的分類水平上, 隨著取樣點的增加, 檢測出的土壤真菌類群的數目呈現出不斷增加的趨勢, 且增速不同, 但逐漸變緩。當取樣點達到82個時, 檢測出的土壤真菌類群的數目達到最大, 這說明當取樣點數較少時, 可能存在較多的真菌類群沒有被檢測出來的情況。特別是在OTU的水平上, 當樣品數量達到82個時, 真菌OTU數量仍呈現出較快增長的趨勢, 由此推測, 如果樣品數量增加較多, 被檢測出來的真菌OTU數量還會有較大增加, 今后的試驗進一步在木論喀斯特常綠落葉闊葉混交林中, 探究取樣數目與土壤真菌類群數目在屬和OTU水平上的聯系。在以往對草地、農田或是在森林等土壤微生物研究中, 均應重視取樣數量對于研究結果的影響[33-35]。取樣數量除了取決于研究目的和研究深度外, 實驗研究成本也會對其產生一定影響, 因為高通量測序單個樣品所需經費相對較高, 因此, 我們在實際研究當中應對取樣數量進行權衡, 既要能滿足研究目的、反應研究對象的微生物多樣性水平, 又要能兼顧到研究費用。除此之外, 檢測出的真菌類群也會受到文庫的構建和測序深度的選擇等因素的影響。因此, 庫容和測序深度的增加, 也有可能檢測出來更多的真菌類群。還有, 測序區間的選擇也是影響研究結果的一個因素, 例如: 測序的高變區不同, 其分類信息的精確性也會不一樣。
4 結論
木論喀斯特常綠落葉闊葉混交林土壤真菌分屬于6門32綱126目336科886屬。相對豐度最高的2個門分別是子囊菌門、接合菌門, 其所占比例總和達到了92.22%; 綱類水平下, 以接合菌綱最高, 其次是座囊菌綱和糞殼菌綱, 三者所占比例總和達到了64.09%; 目類水平下, 相對豐度最高的是被孢霉目, 其占比為22.65%, 其余各目所占比例均低于10%; 相對豐度最高的5個科分別是被孢霉科、格孢菌科、發菌科、炭角菌科和古生菌科, 其所占比例為14.59%; 相對豐度最高的5個屬分別是被孢霉屬、小畫線殼屬、白粉菌屬、周刺座霉屬和青霉屬, 其所占比例總和為9.28%。
隨著采樣點數量的增加, 不同分類土壤真菌水平有不同程度的增加, 在門、綱、目、科的分類水平上, 1個、15個、48個、73個樣品基本上可代表全部樣品檢測出來不同分類水平的細菌多樣性信息; 而屬和OUT數仍隨樣品數量增加而增加, 未呈現平緩趨勢, 說明在該生境內, 繼續增加取樣數目, 屬和OTU數目還會有明細增加, 今后可通過補充采樣來進一步了解該森林土壤真菌屬和OUT特征。
致謝:感謝陳莉、胡芳同學在野外土樣采集中的幫助, 感謝木論國家級自然保護區工作人員在野外工作中的支持和幫助。
[1] DEQUIEDT S, SABY N P A, LELIEVRE M, et al. Biogeographical patterns of soil molecular microbial biomass as influenced by soil characteristics and management[J]. Global Ecology and Biogeography, 2011, 20(4): 641–652.
[2] 宋長青, 吳金水, 陸雅海, 等. 中國土壤微生物學研究10年回顧[J]. 地球科學進展, 2013, 28(10): 1087–1105.
[3] 賀紀正, 王軍濤. 土壤微生物群落構建理論與時空演變特征[J]. 生態學報, 2015, 35(20): 6575–6583.
[4] 盛玉鈺, 叢靜, 盧慧, 等. 神農架國家公園林線過渡帶土壤真菌多樣性[J]. 生態學報, 2018, 38(15): 5322–5330.
[5] TEDERSOO L, BAHRAM M, POLME S, et al. Global diversity and geography of soil fungi[J]. Science, 2014, 346,10.1126/science.1256688 .
[6] 陳曉, 劉勇, 李國雷, 等. 土壤真菌研究方法及人為干擾對森林土壤真菌群落影響研究進展[J]. 世界林業研究, 2011, 24(5): 7–12.
[7] 王楠楠, 楊雪, 李世蘭, 等. 降水變化驅動下紅松闊葉林土壤真菌多樣性的分布格局[J]. 應用生態學報, 2013, 24(7): 1985–1990.
[8] VORISKOVA J, BRABCOVA V, CAJTHAML T, et al. Seasonal dynamics of fungal communities in a temperate oak forest soil[J]. The New phytologist, 2014, 201(1): 269–278.
[9] XIAO Wenya, ZHAO Jiahao, YAN Xinlin, et al. Tree Diversity Determines the Diversity of the Taxonomic and Functional Structure of the Fungal Community in Forest Litter in Southern China[J]. Forest Science, 2019, 65(1): 40–47.
[10] 張騰升. 亞熱帶不同植被恢復階段植物與土壤微生物多樣性特征及其相關關系[D]. 南昌: 南昌工程學院, 2019.
[11] 隋心, 楊立賓, 崔福星, 等. 湯旺河國家公園紅松林土壤真菌多樣性研究[J]. 中國農學通報, 2019, 35(22): 84–90.
[12] 喬沙沙, 周永娜, 柴寶峰, 等. 關帝山森林土壤真菌群落結構與遺傳多樣性特征[J]. 環境科學, 2017, 38(6): 2502– 2512.
[13] 吳佳偉, 楊瑞, 王勇, 等. 貴州草海流域三種不同植被類型根際土壤真菌結構組成和多樣性[J]. 菌物學報, 2020, 39(7): 1250–1262.
[14] 周玉杰, 李建華, 張廣宇, 等. 基于高通量測序的橡膠林土壤真菌多樣性及群落組成分析[J]. 南方農業學報, 2018, 49(9): 1729–1735.
[15] 葉文雨, 謝序澤, 許鈺瀅, 等. 基于高通量測序技術分析2種菌草根際土壤真菌群落多樣性[J]. 熱帶作物學報, 2020, 41(3): 556–563.
[16] 鄧嬌嬌, 朱文旭, 張巖, 等. 遼西北風沙區不同人工林土壤真菌群落結構及功能特征[J]. 林業科學研究, 2020, 33(1): 44–54.
[17] 武俊男, 劉昱辛, 周雪, 等. 基于Illumina MiSeq測序平臺分析長期不同施肥處理對黑土真菌群落的影響[J]. 微生物學報, 2018, 58(9): 1658–1671.
[18] 陳莉, 宋同清, 王華, 等. 木論喀斯特常綠落葉闊葉混交林土壤細菌多樣性及其最優采樣數[J]. 生態學報, 2019, 39(09): 3287–3296.
[19] DU Hu, HU Fang, ZENG Fuping, et al. Spatial distribution of tree species in evergreen-deciduous broadleaf karst forests in southwest China[J]. Scientific reports, 2017, 7(1): 15664.
[20] 蘭斯安, 宋敏, 曾馥平, 等. 木論喀斯特森林木本植物多樣性垂直格局[J]. 生態學報, 2016, 36(22): 7374–7383.
[21] 毛海萍, 袁開, 金仁耀, 等. 基于傳統分離培養和高通量測序分析市售咸鰳魚中微生物多樣性[J]. 食品研究與開發, 2019, 40(21): 193–201.
[22] MAGOC T, SALZBERG S L. FLASH: fast length adjustment of short reads to improve genome assemblies[J]. Bioinformatics (Oxford, England), 2011, 27(21): 2957–2963.
[23] CAPORASO J G, KUCZYNSKI J, STOMBAUGH J, et al. QIIME allows analysis of high-throughput community sequencing data[J]. Nature Methods: Techniques for life scientists and chemists, 2010, 7(5): 335–336.
[24] EDGAR R C. UPARSE: highly accurate OTU sequences from microbial amplicon reads[J]. Nature methods, 2013, 10(10): 996–998.
[25] STACKEBRANDT E, GOEBEL B M. Taxonomic Note: A Place for DNA-DNA Reassociation and 16S rRNA Sequence Analysis in the Present Species Definition in Bacteriology[J]. International Journal of Systematic Bacteriology, 1994, 44(4): 846–849.
[26] MAIDAK B L, COLE J R, LILBURN T G, et al. The RDP-II (Ribosomal Database Project)[J]. Nucleic acids research, 2001, 29(1): 173–174.
[27] 趙愛花, 杜曉軍, 臧婧, 等. 寶天曼落葉闊葉林土壤細菌多樣性[J]. 生物多樣性, 2015, 23(5): 649–657.
[28] O'BRIEN H E, PARRENT J L, JACKSON J A, et al. Fungal community analysis by large-scale sequencing of environmental samples[J]. Applied and environmental microbiology, 2005, 71(9): 5544–5550.
[29] LIENHARD P, TERRAT S, PREVOST-BOURE N C, et al. Pyrosequencing evidences the impact of cropping on soil bacterial and fungal diversity in Laos tropical grassland[J]. Agronomy for Sustainable Development, 2014, 34(2): 525–533.
[30] LIM Y W, KIM B K, KIM C M, et al. Assessment of soil fungal communities using pyrosequencing[J]. Journal of microbiology (Seoul, Korea), 2010, 48(3): 284–289.
[31] FIERER N, JACKSON R B. The diversity and biogeo-graphy of soil bacterial communities[J]. Proceedings of the National Academy of ences of the United States of America, 2006, 103(3): 626–631.
[32] LI Hui, YE Dandan, WANG Xugao, et al. Soil bacterial communities of different natural forest types in Northeast China[J]. Plant and Soil, 2014, 383(1/2): 203–216.
[33] GRIFFITHS R I, THOMSON B C, JAMES P, et al. The bacterial biogeography of British soils[J]. Environmental Microbiology, 2011, 13(6): 1642–1654.
[34] JONES R T, ROBESON MS, LAUBER C L, et al. A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses[J]. The ISME journal, 2009, 3(4): 442– 453.
[35] LAUBER L C, HAMADY M, KNIGHT R, et al. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale[J]. Applied and environmental microbiology, 2009, 75(15): 5111–5120.
Soil fungal community composition in the Mulun karst evergreen and deciduous broad-leaved mixed forest
GU Junkun1,2, ZENG Fuping1,2, SONG Tongqing1,2, PENG Wanxia1,2, SU Liang1,2, DU Hu1,2,*
1. Key Laboratory of Agro-ecological Processes in Subtropical Region, Institute of Subtropical Agriculture, Chinese Academic of Sciences, Changsha, Hunan 410125, China 2. Guangxi Key Laboratory of Karst Ecological Processes and Services, Huanjiang Observation and Research Station of Karst Ecosystem, Huanjiang, Guangxi 547100, China 3. College of Resources and Environment, Hunan Agricultural University, Changsha 410125, China
Soil microbes play essential roles in forest ecosystems. To understand the diversity of soil fungal in karst forest, we investigated soil fungal community structure in the Mulun karst evergreen and deciduous broad-leaved mixed forest using the technology of Illumina Miseq. The results showed that in the 82 soil sampling sites, the number of fungal taxa detected at different classification levels increased with the increasing number of sampling points. When all 82 samples were considered, the number of relative fungal groups include 6 phyla, 32 classes, 126 orders, 336 families, 886 genera, and 132616 operational taxonomic units (OTUs). The mean values of relative fungal taxa in the 82 samples were 5.41 phyla, 22.13 classes, 71.09 orders, 144.89 families, 216.66 genera, and 1256.44 OTUs. At the classification level of phylum, class, and order, the dominant groups were(50.09%),(34.54%),(22.65%), respectively. These preliminary results show that the soil has a relatively high level of fungal diversity in the karst evergreen and deciduous broad-leaved mixed forest, which lays the foundation for further studies on the relationship between soil fungal diversity and plant diversity and other related scientific questions.
soil fungal diversity; high-throughput sequencing; number of samples; forest; karst ecosystem
谷俊錕, 曾馥平, 宋同清, 等. 木論喀斯特常綠落葉闊葉混交林土壤真菌群落多樣性特征[J]. 生態科學, 2022, 41(3): 54–61.
GU Junkun, ZENG Fuping, SONG Tongqing, et al. Soil fungal community composition in the Mulun karst evergreen and deciduous broad-leaved mixed forest[J]. Ecological Science, 2022, 41(3): 54–61.
10.14108/j.cnki.1008-8873.2022.03.006
S154.38
A
1008-8873(2022)03-054-08
2020-07-16;
2020-09-08
廣西重點研發計劃項目(桂科AB17129009); 國家自然科學基金項目(31770495, 31870712, 31971487); 河池市特聘專家項目
谷俊錕(1997—), 男, 湖南張家界人, 碩士, 主要從事生態學研究。E-mail: 505218176@qq.com
杜虎(1986—), 男, 副研究員, 主要從事喀斯特生物多樣性研究。E-mail: hudu@isa.ac.cn
猜你喜歡
西北民族大學學報(自然科學版)(2021年4期)2021-12-29 02:54:24
數學小靈通(1-2年級)(2021年4期)2021-06-09 06:25:56
大眾健康(2021年6期)2021-06-08 19:30:06
小聰仔(科普版)(2020年12期)2021-01-18 09:16:52
東方少年·布老虎畫刊(2020年4期)2020-06-08 15:48:10
學生天地(2019年32期)2019-08-25 08:55:22
中學生數理化·七年級數學人教版(2019年4期)2019-05-20 10:06:32
中學生數理化·七年級數學人教版(2018年6期)2018-06-26 08:36:06
小天使·一年級語數英綜合(2017年11期)2017-12-05 18:49:56
初中生世界·七年級(2017年9期)2017-10-13 22:27:46
