線粒體腦肌病的MRI診斷
——2022年讀片窗(5)
2022-05-21 08:52:50王龍勝
安徽醫學 2022年5期
關鍵詞:信號
王龍勝
作者單位: 230601 安徽合肥 安徽醫科大學第二附屬醫院放射科
1 病史摘要
患者,女性,55歲,1周前患者無明顯誘因下出現胡言亂語、不認識家人、理解不能,無四肢無力、頭暈頭痛、飲水嗆咳,癥狀進行性加重。病程中患者飲食睡眠可,大小便正常,近期體質量未見明顯下降。既往有糖尿病病史。體格檢查:體溫36.0℃ 、脈搏83次/分、 呼吸20次/分、 血壓 124/92 mmHg (1 mmHg≈0.133 kPa),神清,精神一般,反應遲鈍,自言自語,問話不答,雙側瞳孔等大同圓,四肢肌力檢查不配合,能自行行走。病理征不配合,病程中患者飲食睡眠可,大小便正常,近期體質量未見明顯下降。實驗室檢查:C-反應蛋白<0.5 mg/L,白細胞計數6.71×109/L,血紅蛋白142 g/L,血小板計數274×109/L,血漿乳酸2.9 mmol/L↑。腦脊液生化:葡萄糖7.26 mmol/L↑,氯122.8 mmol/L,腺苷脫氨酶1 U/L,腦脊液總蛋白483 mg/L↑。 腦脊液常規:潘氏試驗弱陽性↑,白細胞35×106/L↑。腦脊液墨汁染色未檢出新型隱球菌。腦脊液乳酸:乳酸3.8 mmol/L↑。
2 MRI檢查所見
雙側枕葉可見對稱斑片狀長T1、長T2信號(圖1、2),FLAIR序列呈高信號(圖3),DWI序列呈高信號(圖4),ADC圖呈稍低信號(圖5),測ADC值約0.76×10-3mm2/s;腦室系統擴大,腦溝裂明顯增寬,中線結構居中,幕下小腦及腦干未見明顯異常信號。MRS右側枕葉病灶顯示NAA峰及CHO峰稍下降,可見明顯峰倒置的Lac(圖6)。

圖1 T1WI

圖2 T2WI

圖3 FLAIR
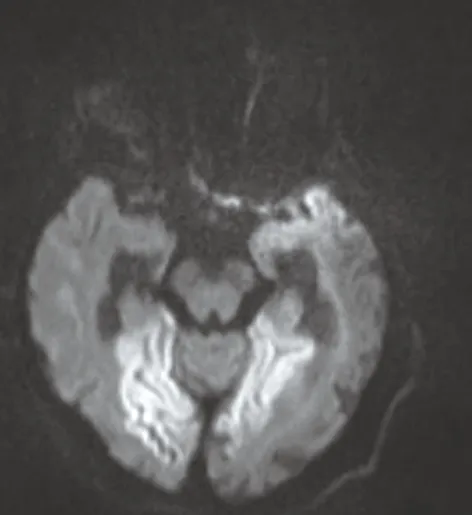
圖4 DWI

圖5 ADC

圖6 MRS
3 臨床診斷
線粒體DNA高敏感測序分析結果陽性,臨床診斷為線粒體腦肌病。
4 討論
線粒體腦肌病是由于線粒體遺傳物質DNA有缺陷引起的一組與線粒體氧化磷酸化功能異常有關的遺傳代謝性疾病,有多種類型,包括肌陣攣性癲伴破碎紅纖維綜合征(myoclonus epilep- sy with ragged-red fibers,MERRF)、線粒體腦肌病伴高乳酸血癥及卒中樣發作、慢性進行性眼外肌癱瘓綜合征(chronic progressive external othalmoplegia,CPEO)、Leigh綜合征(leigh syndrome,LS)、Kearne-Sayre綜合征(Kearne-Sayre syndrome,KSS)等,其中線粒體腦病是最常見的一種。它是一種慢性進行性的神經退行性疾病,腦部病理改變為腦組織海綿樣變性、灶狀壞死、脫髓鞘伴鐵沉積、膠質細胞增生等。
臨床特點:①該病多見于兒童青少年,以 5~15歲最好發,也可見于成人,本例患者是55歲中年女性;②臨床表現復雜多樣,缺乏特異性,多以間斷性頭痛、嘔吐、智力障礙、肌力異常、癲癇發作等為首發癥狀,且癥狀進行性加重;③血和腦脊液檢查,乳酸和丙酮酸增高,本例血和腦脊液乳酸檢查均升高;④腦電圖、肌電圖檢查,表現異常,呈癲癇表現,肌源性或神經源性損害;⑤肌肉活檢:可見線粒體異常和不整紅邊纖維(RRF);⑥基因檢測:mtDNA中tRNA亮氨酸(Leu)基因核甘4243位點發生A-G點突變。
MRI表現:①發病部位及病變形態,好發于頂枕葉、顳葉的腦皮層及皮層下區,病變分布與腦動脈供血區不一致;病變形態不規則呈斑片、條狀,病變區灰白質界限欠清楚;②信號特點:呈長T1長T2信號,FLAIR及DWI 均呈高信號,ADC值降低, PWI示病變區呈高灌注狀態;③病灶數目:單發或多發對稱性病灶,可新舊病灶并存,具有此起彼伏、游走的特點;④局部腦萎縮:陳舊病灶區腦皮質變薄,腦溝裂增寬,呈萎縮樣改變;⑤增強掃描:病灶無強化,或輕度腦回樣強化;⑥MRS:具有特征性,病變區表現NAA降低和Lac升高, NAA/Cr降低,Lac/Cr升高;另外,腦脊液區1H-MRS也可出現Lac峰。周志凌等[1]報道13例線粒體腦病中10例患者腦脊液區可見Lac峰,有助于對腦脊液乳酸水平的檢測;⑦MRA:表現大腦中、后動脈分支的小血管增多,但罕有大血管閉塞或狹窄。
鑒別診斷:線粒體腦肌病的病變形態和信號與腦梗死和腦炎[2]相似,容易混淆需要鑒別。①急性腦梗死:發病急,病變部位與腦血管供血范圍一致,腦內血管多有不同程度血管閉塞或狹窄改變,增強掃描常有明顯腦回樣強化;而線粒體腦肌病病變部位與血管供應范圍不一致、且多發病變常不在同一血管支配區,病變可在短期內好轉或進展,具有多灶性、對稱性、游走的特性,增強掃描病灶多不強化或僅見輕度沿病變腦回分布的線樣強化。②腦炎:臨床常有發熱、頭痛,甚至昏迷等癥狀,腦脊液檢查也具有其表現特點,患者多有感染史,主要累及顳葉和邊緣系統。
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
