Ru 摻雜對Pt(111)面CO 覆蓋度影響的理論研究
2022-06-08 07:09:22丁浩然姚陳忠
長治學(xué)院學(xué)報 2022年2期
關(guān)鍵詞:催化劑
丁浩然,姚陳忠
(運城學(xué)院 應(yīng)用化學(xué)系,山西 運城 044000)
引言
能源供需不平衡、化石燃料面臨耗竭和環(huán)境污染等問題是當(dāng)今人類社會面臨的主要挑戰(zhàn)。因此,尋求更高效、更安全、更清潔甚至“ 零排放” 的新能源技術(shù),是實現(xiàn)人類可持續(xù)發(fā)展的關(guān)鍵[1-2]。直接甲醇燃料電池(DMFC)是一種將甲醇中儲存的化學(xué)能直接轉(zhuǎn)化為電能的潔凈環(huán)保器件,因其不受卡諾循環(huán)的限制,兼具高轉(zhuǎn)換效率與低廢物排放等優(yōu)點,得到研究者越來越多的關(guān)注[1-3]。
DMFC 的關(guān)鍵組成部分之一是陽極甲醇氧化反應(yīng)的電催化劑。甲醇氧化反應(yīng)由于其六電子轉(zhuǎn)移過程而在動力學(xué)上速度緩慢[3-4]。Pt 基催化劑由于其優(yōu)異的甲醇電催化性能,是目前研究的熱點。研究發(fā)現(xiàn),Pt 的催化活性與顆粒大小、形貌、結(jié)構(gòu)和表面狀態(tài)密切相關(guān),特別是高的CO 覆蓋度將會導(dǎo)致催化劑快速失活[5]。為了解決這一問題,一種策略是開發(fā)新型Pt 基雙金屬材料,其中Pt 作為甲醇氧化活性中心,另一金屬成分作為CO 去除劑。利用這種協(xié)同效應(yīng),該催化劑同時對甲醇氧化反應(yīng)表現(xiàn)出良好的活性和優(yōu)良的耐久性。其中,Pt-Ru 雙金屬納米材料被認(rèn)為是迄今為止性能最好的抗CO 中毒催化劑[6-7]。
基于此,文章首先通過理論計算,系統(tǒng)研究了Ru 摻雜對Pt 電子結(jié)構(gòu)的影響;隨后計算摻雜前后CO 吸附能和CO 活化轉(zhuǎn)化的差異;最終通過理論計算解釋Ru 摻雜能夠降低Pt 催化劑表面CO 覆蓋度的原因。
1 計算方法和模型
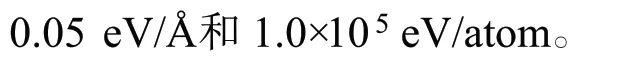
因為X 射線衍射儀表征結(jié)果顯示Pt(111)面是金屬Pt 的主要暴露晶面[12],所以計算使用Pt(111)面。Pt(111)面采用的4 層周期性模型,真空層厚度為1.5 nm。計算時,固定Pt(111)面的最下兩層,其余原子和吸附分子可以弛豫。我們考慮了不同吸附位的吸附(圖1):頂位(top),橋位(bridge),三重穴位hcp 和fcc,在文中只比較最穩(wěn)定的吸附位。
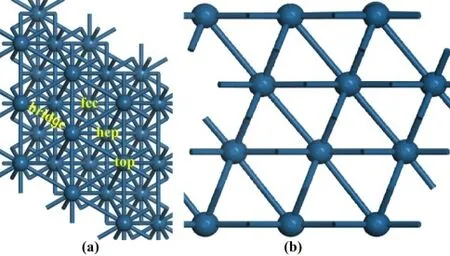
圖1 Pt(111)面的俯視(a)和主視圖(b)
吸附能的計算公式是:Eads= E(mol)+ E(slab)-E(mol/slab),其中E(mol),E(slab)和E(mol/slab)分別為自由分子的能量、底物的能量和吸附物吸附在底物上時系統(tǒng)的總能量。因此,吸附能越大說明吸附物和底物之間的相互作用力就越大。
2 結(jié)果與討論
首先筆者研究Ru 摻雜對Pt(111)面電荷結(jié)構(gòu)的影響。因為一個Ru 摻雜在最上層,所以我們只討論第一層原子的電子結(jié)構(gòu)。摻雜前,bader 電荷顯示最上層每個Pt 的電子為-0.06 e,這說明在Pt(111)表面有部分電子的聚集。摻雜后,Ru 的bader 電荷為0.46 e,Pt 的電荷約為-0.11 e。bader電荷結(jié)果顯示,摻雜后Ru 的電子向Pt 轉(zhuǎn)移。這是因為Ru 和Pt 的電負(fù)性為1.54 和1.72,后者的電負(fù)性大于前者,所以Ru 的電子向Pt 轉(zhuǎn)移。
圖2 是Pt(111)和Ru-Pt(111)面第一層原子的分波態(tài)密度(PDOS)。比較圖2(a)和圖2(b)可以看到,由于摻雜量較低,Pt 的PDOS 圖沒有明顯變化。然而,Pt(111)面的d 帶中心遠(yuǎn)離費米能級。這是因為摻雜Ru 后,表面Pt 原子的bader 電荷增加,因此其d 帶中心從-2.08 eV 遷移到-2.13 eV。因為Ru 原子的d 帶中心位于-1.28 eV(圖2c),所以造成摻雜后第一層所有原子的d 帶中心又靠近費米能級(-2.13 vs. -2.04 eV)。總體來說,Ru 原子摻雜可以使表面原子的d 帶中心靠近費米能級,從而可能對提高催化劑的穩(wěn)定性有利[13]。
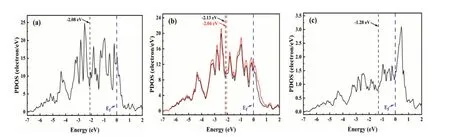
圖2 Pt(111)和Ru-Pt(111)面第一層原子的分波態(tài)密度

首先,我們考慮摻雜前后CO 吸附能的變化。CO 以C 端吸附在Pt(111)面,其最穩(wěn)定吸附位是fcc 位 (圖 3a)。此時,Pt-C 鍵鍵長為0.211 nm,對應(yīng)的吸附能為1.82 eV。考慮熵的貢獻,300 K時CO 的吸附能為1.20 eV。高的吸附能說明CO在常溫時很難從Pt 表面脫附,從而導(dǎo)致DMFC電催化劑極易中毒。摻雜Ru 后,CO 仍以C 端吸附,此時Pt-C 和Ru-C 鍵長分別為0.216 nm 和0.206 nm (圖 3d),吸附能等于1.81 eV。Bader 電荷結(jié)果顯示,摻雜前Pt(111)表面向CO 轉(zhuǎn)移0.39 e;摻雜后表面向CO 轉(zhuǎn)移0.34 e,這說明摻雜抑制了表面電荷的轉(zhuǎn)移。CO 吸附時,盡管摻雜后Ru-C 鍵長小于Pt-C 鍵長,但是Pt-C 鍵長增加,這可能是CO 吸附能沒有變化所致。因此,Ru 的摻雜不能改變CO 在Pt 電極表面的吸附能。
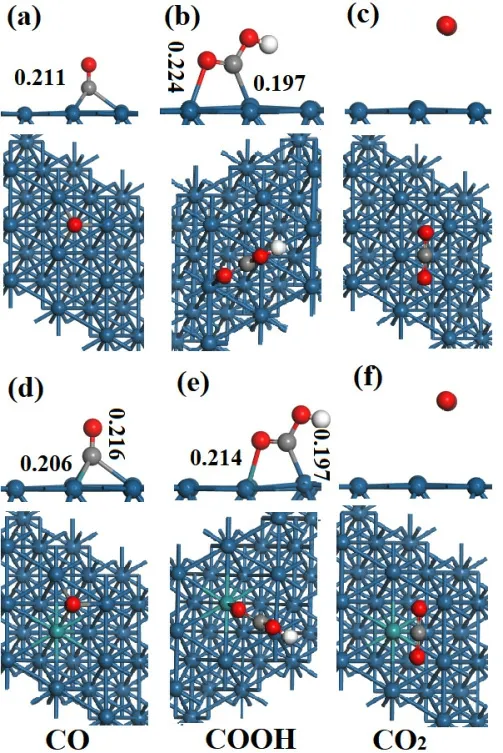
圖3 Pt(111)和Ru-Pt(111)面吸附CO、COOH 和CO2 構(gòu)型圖
COOH 的最穩(wěn)定吸附位是橋位,O 和C 原子分別吸附于相鄰的Pt 原子頂位,OH 指向Pt(111)面 (圖 3b)。Pt-C 和Pt-O 鍵長分別為0.197 nm 和0.224 nm,對應(yīng)的吸附能為2.88 eV。當(dāng)COOH吸附在Ru-Pt(111)面時,其吸附構(gòu)型沒有發(fā)生變化,但是O 傾向于吸附在Ru 位。此時,Pt-C 和Ru-O 鍵長分別為0.197 nm 和0.214 nm (圖 3e),吸附能為3.25 eV。摻雜后,Pt-C 鍵長沒有變化,但是Ru-O 鍵長縮短,這可能是吸附穩(wěn)定性增加的一個原因。Bader 電荷結(jié)果顯示,Pt(111)面吸附COOH 時,Pt 向COOH 轉(zhuǎn)移0.08 e;當(dāng)COOH吸附在Ru-Pt(111)面時,其向COOH 轉(zhuǎn)移0.12 eV。因此,鍵長的減少和電荷轉(zhuǎn)移數(shù)量的增加造成COOH 在Ru-Pt(111)面的吸附增加。
CO2在Pt(111)和Ru-Pt(111)面上吸附時(圖3c 和圖3f),其分子遠(yuǎn)離表面,屬于物理吸附,吸附能分別只有0.08 eV 和0.02 eV。Bader 電荷也顯示CO2吸附前后,二者之間沒有的電荷轉(zhuǎn)移,這與吸附能計算結(jié)果一致。
對于基元反應(yīng)來說,反應(yīng)熱的定義為

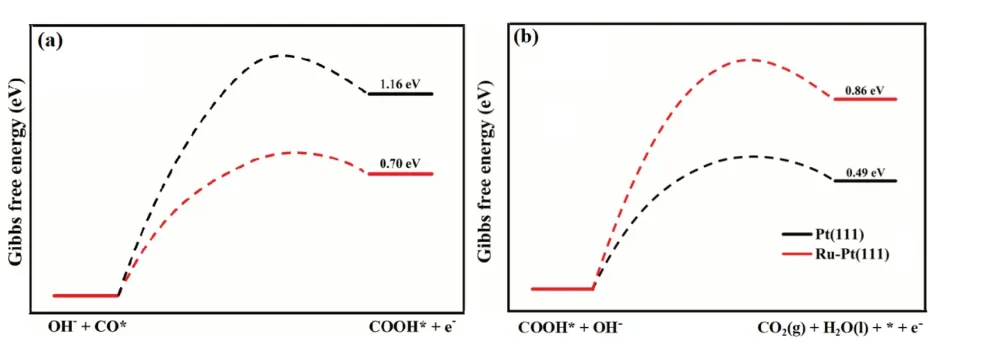
圖4 Pt(111) 和Ru- Pt(111)面OH- + CO* → COOH* + e-和COOH* + OH- → CO2 (g) + H2O (l) + * + e-的反應(yīng)熱(a 圖缺標(biāo)注Pt(111)和Ru-Pt(111)


3 結(jié)論
基于DFT 計算,文章詳細(xì)的討論了Ru 摻雜對CO 在Pt(111)面覆蓋度的影響。我們從兩個方面考慮了降低CO 覆蓋度的方法:對CO 吸附能力和轉(zhuǎn)化活化能的影響。研究結(jié)果如下:
(1)Ru 摻雜后,Ru 核外電子向Pt 轉(zhuǎn)移,使Pt 核外電子富集。同時,Ru 摻雜使電極表面的d 帶中心向費米中心偏移,從而提高其催化活性。
(2)Ru 摻雜不會影響CO 和CO2的吸附能,但是能夠提高COOH 的吸附能。
(3)基于BEP 理論,Ru 摻雜不僅改變了反應(yīng)的速控步驟,同時有利于降低CO 的覆蓋度。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學(xué)學(xué)報(工學(xué)版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(xué)(2015年4期)2016-01-17 09:01:27
應(yīng)用化工(2014年3期)2014-08-16 13:23:50
