QuEChERS前處理結合超高效液相色譜-串聯質譜法同時檢測茶葉中14種農藥殘留
2022-06-11 11:20:16朱穎潔曹燕卿董方霆
食品工業科技 2022年12期
關鍵詞:檢測
朱穎潔,曹燕卿,毛 劼,張 康,董方霆,何 昆,王 娜
(國家生物醫學分析中心,北京 100850)
我國是茶葉的發源地,也是茶葉生產和消費量最大的國家。由于茶樹生長喜溫暖潮濕環境、種植單一化、缺乏生物多樣性的特點,較易發生病蟲害,其規模化種植過程中離不開農藥的使用,農藥殘留問題難以避免[1-2]。國家允許在茶葉上使用一些高效低毒的農藥,在GB 2763-2021《食品安全國家標準 食品中農藥最大殘留限量》中規定了106項茶葉農藥殘留限量標準[3]。然而,由于部分商家追求利益盲目施加農藥和化肥催生、茶農使用農藥不科學等原因,食品安全監督檢查中茶葉農殘超標的情況時有發生,甚至屢有禁用高毒農藥的檢出,嚴重危害消費者安全與市場信心[4-5]。
近年來,農藥殘留分析方法向著快速、簡單、靈敏、低成本、易普及的方向發展。液相色譜-串聯質譜法和氣相色譜-串聯質譜法是最常用的農藥多殘留檢測分析手段,具有高靈敏度、高通量、定量準確等優點[6-9]。由于茶葉樣品基質復雜,含有大量的茶多酚、生物堿、色素和有機酸等物質,檢測過程中會產生較強的基質效應,必須通過前處理凈化以減弱對目標農藥檢測的影響[10-11]。茶葉農藥殘留檢測中傳統的樣品前處理技術如固相萃取[12-13]、液液萃取[14]、基質固相分散萃取[15]等常需要多次凈化步驟,操作較為繁瑣和耗時。2003年美國農業部Anastasiadis等發布的QuEChERS法[16],是液液萃取法與基質分散固相萃取法相結合形成的一種前處理方法,通常使用乙腈或含1%乙酸的乙腈提取,加入無水硫酸鎂(MgSO4)、氯化鈉(NaCl)除水,配合使用N-丙基乙二胺(PSA)、石墨化炭黑(GCB)和十八烷基硅烷(C18)等進一步凈化,對不同種類的農藥均有較好的回收率,具有操作簡單、快速高效、成本低廉的優點[17],在果蔬[18-20]、食用菌[21]、中藥材[22]等植物源食品的多農殘檢測中得到了廣泛的應用。本研究的主要目的是開發一種基于QuEChERS前處理技術結合UPLC-MS/MS,同時檢測茶葉中14種常見農藥殘留的高靈敏、快速檢測方法,考察提取液組分、QuEChERS凈化柱等對回收率的影響,確定了最優條件,并將方法應用于30種市售茶葉樣品檢測,以期為高效和準確地評價茶葉的質量安全提供參考。
1 材料與方法
1.1 材料與儀器
14種農藥標準品(啶蟲脒、涕滅威、克百威、內吸磷、滅線磷、吡蟲啉、茚蟲威、甲胺磷、滅多威、氧化樂果、甲基對硫磷、硫環磷、丙溴磷、噻蟲嗪)(100 mg/L) 天津阿爾塔科技有限公司,于-20 ℃冰箱保存;30種茶葉包括不同種類綠茶(獅峰龍井、竹葉青、采花毛尖、西湖龍井、太平猴魁茶、桐城小花、綠楊春、清明茶高山綠)、紅茶(武夷山紅茶、祁門紅茶、生態野生紅茶、古樹紅茶、金駿眉)、白茶(牡丹王白茶、政和白茶、安吉白茶)、烏龍茶(鐵觀音、大紅袍、老樅水仙、馬頭巖肉桂)、花茶(茉莉花茶、菊花茶、蒲公英玫瑰茶、玫瑰花茶)、普洱茶、青稞茶、刺五加茶等 購于當地超市和茶葉專賣店,西湖龍井和古樹紅茶按照《GB 23200.121-2021》所示LC-MS/MS方法檢測未檢出本研究涉及的14種農藥,分別作為空白綠茶和紅茶基質進行相關研究;QuEChERS鹽包(含有6 g MgSO4,1.5 g乙酸鈉) 北京綠綿科技有限公司;尼龍微孔濾膜(型號SHIMEN DISC,孔徑0.22 μm) 日本SHIMADZU公司;乙腈、甲醇、甲酸 色譜純,美國Fisher公司;甲酸銨、乙酸 分析純,德國Merk公司;4種QuEChERS凈化柱及其使用方法如表1所示。

圖 1 14種農藥的總離子流圖Fig.1 Total ion chromatogram (TIC) of 14 pesticides
Q-Trap 6500質譜儀 美國AB Sciex公司;LC-30AD液相色譜儀 日本SHIMADZU公司;Centrifuge 5810R低溫高速離心機 德國Eppendorf公司;QL-901渦旋儀 美國KYLIN-BELL LAB公司;破壁粉碎機 中國天喜公司。
1.2 實驗方法
1.2.1 樣品前處理 提取:稱取1 g粉碎好的茶葉樣品于50 mL離心管中,加入10 mL超純水,渦旋振蕩5 min,加入10 mL 1%乙酸-乙腈溶液,渦旋振蕩30 s,加入QuEChERS鹽包,渦旋振蕩5 min,以9000 r/min離心5 min,得上清提取液。
凈化:經過上述得到的上清液分別使用4種QuEChERS凈化柱進行下一步凈化處理,具體方法見表1。

表1 4種QuEChERS凈化柱型號、成分及使用方法Table 1 Model, component and usage of four kinds of QuEChERS purification columns
1.2.2 溶液配制 混合標準溶液:取14種農藥標準品溶液(100 mg/L)各100 μL于10 mL容量瓶,加入乙腈定容,得到濃度為1 mg/L的混合標準溶液,于-20 ℃冰箱保存。
空白基質溶液:稱取1 g(精確至0.001 g)空白綠茶/紅茶樣品,按照1.2.1節進行提取和凈化,得到相應的空白綠茶/紅茶基質溶液,將該溶液置于-20 ℃冰箱中保存,備用。
基質混合標準溶液1:用空白基質溶液將混合標準溶液稀釋得到0.001、0.005、0.01、0.02、0.05、0.1、0.5、1、2、5、10 μg/L等系列質量濃度的基質混合標準工作溶液,用于確定方法檢出限和定量限。
基質混合標準溶液2:用空白基質溶液將混合標準溶液逐級稀釋得到質量濃度為1、5、10、20、50、100 μg/L的系列基質混合標準工作溶液,用于做標準工作曲線,即基質匹配校準曲線。
1.2.3 色譜條件 色譜柱:SHIMAZU Shim-pack Velox Biphenyl C18色譜柱(3 mm×50 mm,2.7 μm)。分別考察乙腈-水、甲醇-水、0.1%甲酸水溶液-甲醇、0.1%甲酸水溶液-甲醇(含2 mmol/L甲酸銨)、0.1%甲酸水溶液-甲醇(含5 mmol/L甲酸銨)四種流動相對14種農藥檢測結果的影響。流動相洗脫程序:0~0.01 min,5% B;0.01~1.0 min,5%~40% B;1.0~3.0 min,40%~85% B;3.0~4.5 min,85%~95% B;4.5~8.0 min,95% B;8.01~10 min,95%~5% B。進樣體積設置為5 μL;流速設置為0.3 mL/min;色譜柱柱溫35 ℃。
1.2.4 質譜條件 離子源:ESI離子源;將14種農藥混合標準溶液通過針泵進樣方式進行質譜方法調諧,在ESI+和ESI-模式下進行掃描,優化各化合物的質譜條件;檢測方式:多反應監測模式(multiple reaction monitoring,MRM);離子源電壓:5500 V;離子源溫度:500 °C;氣簾氣:35 psi;霧化氣:65 psi;輔助加熱氣:55 psi。
1.2.5 方法學考察
1.2.5.1 基質效應 茶葉的基質較復雜,可能存在基質抑制或基質增強效應。分別用廠家1復雜基質凈化柱處理所得的空白綠茶基質和1%乙酸-乙腈提取溶劑配制濃度為50 μg/L的14種混合農藥標準溶液,重復測定3次,考察14種農藥在綠茶中的基質效應。
基質效應(Matrix effect,ME)按下式計算:

式中:ME表示基質效應,%;Amatrix表示基質混合標準溶液中目標分析物的峰面積(A,peak area);Aextraction表示提取溶劑混合標準溶液中目標分析物的峰面積。
1.2.5.2 方法的線性范圍、檢出限、定量限 采用基質匹配標準溶液建立標準曲線。按照1.2節描述配制質量濃度為1、5、10、20、50、100 μg/L的系列基質混合標準工作液,以各個農藥的峰面積為縱坐標,相應的濃度為橫坐標,獲得目標物農藥的線性回歸方程,分別以信噪比(S/N)為3和10時對應的空白樣品添加濃度為檢出限(Limit of detection, LOD)和定量限(Limit of quantification, LOQ)。
1.2.5.3 回收率和精密度 分別在空白綠茶和紅茶樣品中添加低、中、高濃度(100、500、800 μg/kg)農藥進行加標回收率試驗,各濃度平行試驗6次,計算平均回收率及測定結果的相對標準偏差(RSD)。
1.2.5.4 實際茶樣品檢測 利用本文方法對市售的30種茶葉進行檢測,分析其中14種目標農藥的殘留濃度。
1.3 數據處理
分別利用Analyst 1.7和Sciex OS 1.7.0軟件進行數據采集及處理,Origin 9.0進行數據分析。
2 結果與分析
2.1 色譜、質譜條件優化
使用SHIMAZU Shim-pack Velox Biphenyl C18色譜柱(3 mm×50 mm,2.7 μm),設定流速為0.3 mL/min,分別考察了以乙腈-水、甲醇-水、0.1%甲酸水溶液-甲醇、0.1%甲酸水溶液-甲醇(含2 mmol/L甲酸銨)、0.1%甲酸水溶液-甲醇(含5 mmol/L甲酸銨)為流動相對14種農藥檢測結果的影響,梯度洗脫程序見1.2.3。結果表明,選用0.1%甲酸水溶液-甲醇(2 mmol/L甲酸銨)為流動相時,14種農藥的響應值和峰形最好。14種農藥的總離子流圖見圖1。
將14種農藥的0.1 mg/L混合標準溶液通過針泵進樣方式進行質譜方法調諧,在ESI+和ESI-模式下進行掃描。結果顯示,14種農藥的母離子均為[M+H]+模式。手動調諧優化14種農藥母離子和子離子,選取兩組靈敏度最佳的離子對作為定量離子對和定性離子對,獲得最佳的碰撞電壓和碰撞能量,最終得到MRM模式下14種農藥優化的質譜采集參數如表2所示。
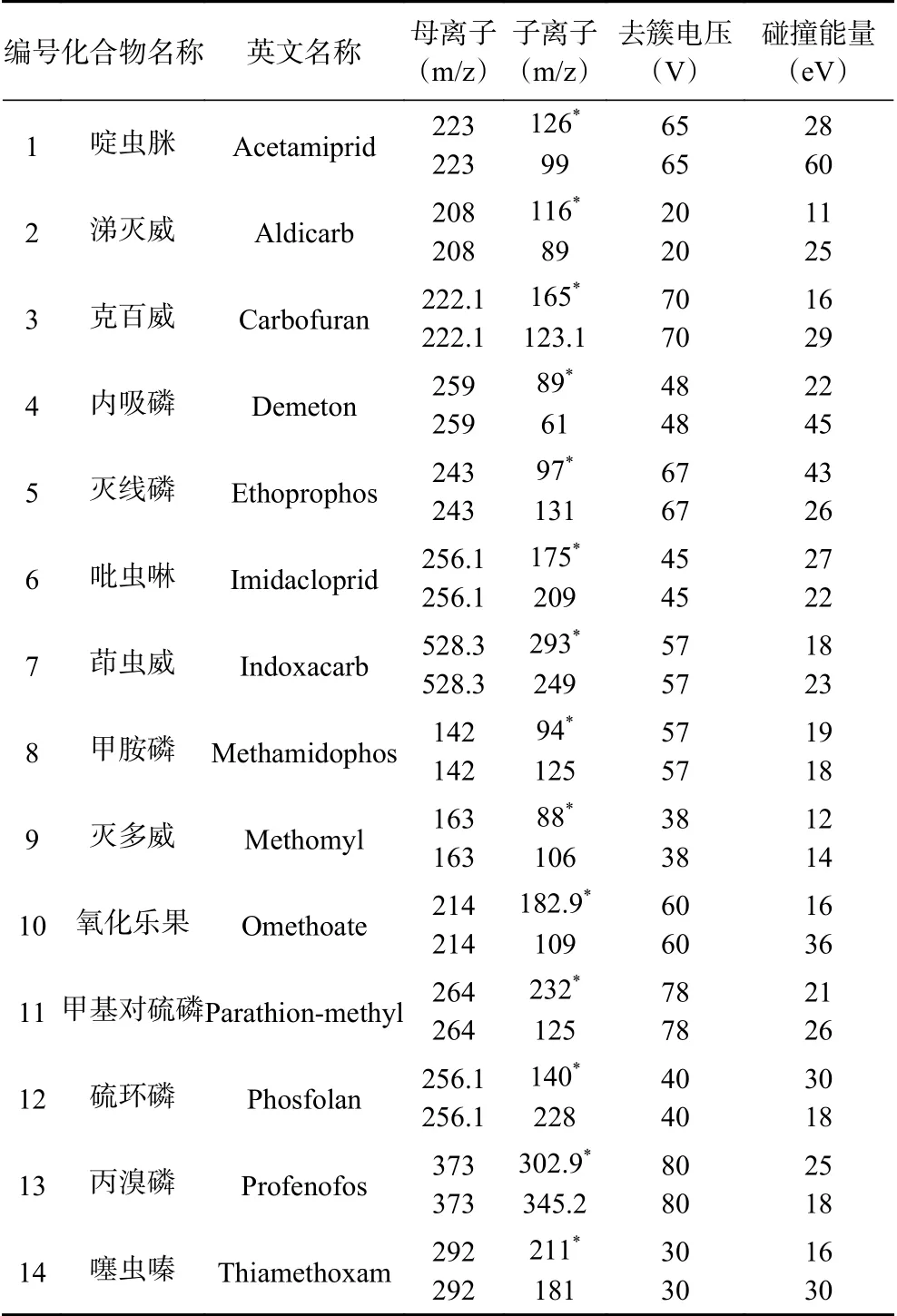
表2 14種農藥的質譜參數Table 2 MS parameters of the 14 pesticides
2.2 提取方法選擇
QuEChERS樣品前處理主要由提取和凈化兩部分組成。在提取階段,常用的提取溶劑有乙腈、丙酮、乙酸乙酯、甲醇、正己烷等。乙腈因最適宜于提取各種極性的農藥殘留,同時不會萃取出很多油性物質(如蠟、油性色素、脂肪等),且僅需加入鹽便可實現與水相的分離,是目前使用最廣泛的提取溶劑。研究表明,在乙腈中加入1%乙酸,并在鹽析過程中加入MgSO4和緩沖鹽CH3COONa可對絕大多數農藥殘留得到較高的回收率[23]。另外,對于含水量低的樣品,通過先向樣品中添加一定量的水,可弱化待分析物與樣品基質之間的相互作用,使待分析物更易在萃取/分配過程中被充分提取[24]。
本研究首先向茶葉樣品中加入適量水充分浸潤,再分別使用乙腈及含1%乙酸的乙腈作為提取溶劑,震蕩均勻后加入含6 g MgSO4和1.5 g CH3COONa的鹽包,進行提取效果的比較,結果表明(圖2),采用1%乙酸-乙腈作為提取溶劑時,其農藥回收率高于乙腈作為提取溶劑,因此,本文選擇1%乙酸-乙腈作為提取溶劑。
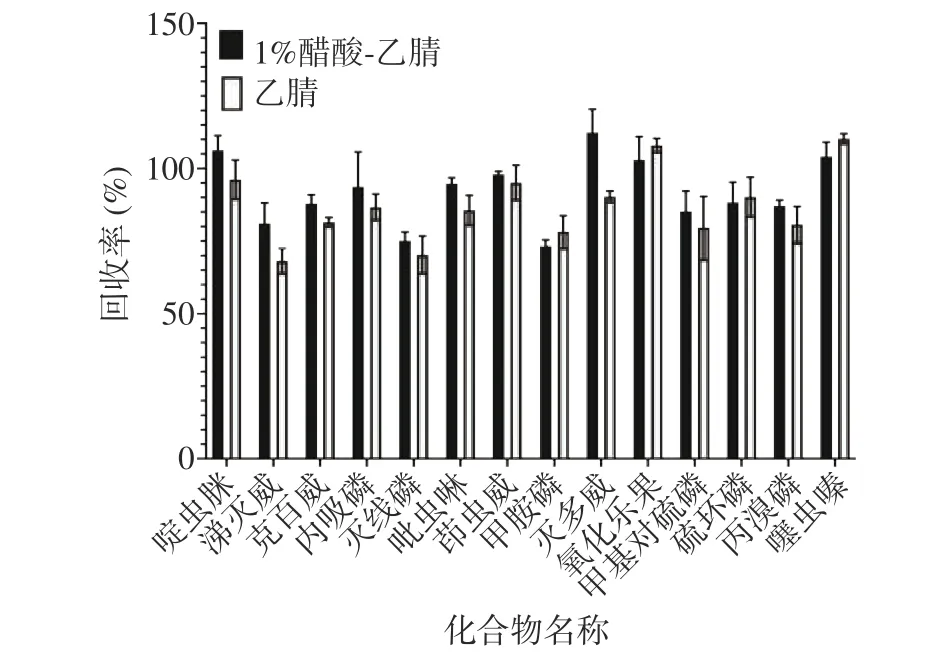
圖 2 不同提取液對目標化合物回收率的影響Fig.2 Effect of different extracts on target compound recovery
2.3 凈化方式優化
茶葉樣品基質復雜,其中的茶多酚、咖啡因、色素、有機酸等物質易和待測目標物共提取,干擾檢測的準確性和靈敏度[6]。常見的凈化劑有無水MgSO4、PSA、GCB和C18等。無水MgSO4能除去提取液的少量水分,PSA可有效去除脂肪酸、有機酸、一些極性色素和糖類,GCB能有效去除具有平面結構的甾醇和色素類雜質,C18吸附劑對油脂的去除效果十分明顯[25]。另外,一些新型凈化材料如多壁碳納米管(MWCNTs)和石墨烯等因被證實能有效去除基質中色素類、糖類、氨基酸等干擾物,并在一定程度上改善GCB對部分平面結構化合物的吸附問題,也逐漸得到應用[26-27]。傳統的凈化方法是向提取液中加入一定量吸附劑,充分震蕩使吸附劑與樣品充分反應,最后離心去除吸附劑。為了使操作更加快速高效,本文使用將凈化劑固相化的QuEChERS凈化柱對提取液進行凈化,省去震蕩、渦旋和離心等步驟從而縮短了樣品前處理時間。
本文考察了在空白綠茶和紅茶樣品中添加800 μg/kg目標化合物,采用兩個廠家四種商品化QuEChERS凈化柱(成分及含量見表1)對茶葉提取液凈化效果和農藥回收率的影響。如圖3所示,由茶葉提取液經不同凈化柱凈化后的顏色深淺可初步判斷,復雜基質凈化柱對于色素雜質的凈化效果好于簡單基質凈化柱,這是因為復雜基質凈化柱中的GCB或MWCNTs對色素有很好的吸附作用。根據圖4可知,對于綠茶樣品,廠家1簡單基質凈化柱對14種農藥的回收率(56.3%~91.1%)整體低于廠家1復雜基質凈化柱(73.4%~112.4%),且色譜圖中雜峰較明顯(見圖5),可見含有GCB(5 mg)的復雜基質凈化柱能有效吸附色素等雜質,降低提取液中高濃度雜質對目標分析物檢測的影響;綠茶樣品經廠家2簡單基質凈化柱處理后對14種農藥回收率(42.7%~79.3%)和經廠家2茶葉專用柱凈化后對14種農藥回收率(52.7%~71.2%)整體低于廠家1凈化柱,說明廠家2兩種凈化柱中較高含量的MWCNTs(分別為15和45 mg)對目標化合物產生了一定吸附從而導致回收率降低。對于紅茶樣品,采用四種凈化柱處理后14種農藥回收率整體高于綠茶樣品,且凈化后樣品顏色比綠茶淺,說明綠茶和紅茶在茶多酚、生物堿、色素等成分上的差異會影響凈化效果和檢測結果,在實際檢測中應根據樣品具體情況進行優化。

圖 3 綠茶和紅茶經不同凈化柱處理后茶葉提取液的顏色對比Fig.3 Comparison of green tea and black tea extracts using different purification columns

圖 4 不同凈化柱對目標化合物回收率的影響Fig.4 Effect of different purification columns on the target compound recovery

圖 5 總離子流色譜圖Fig.5 Total ion chromatograms (TIC)
綜合考慮,本文使用凈化效果和回收率都較好的廠家1復雜基質凈化柱,進一步考察方法的基質效應、線性關系、回收率等,并對實際樣品進行檢測。
2.4 基質效應
茶葉中的茶多酚、生物堿、色素等易和目標物分析物共提取,難以通過凈化全部去除,從而對目標分析物的離子化效率產生影響,即產生基質效應(Matrix effect,簡稱ME)[28-29]。我們分別用廠家1復雜基質凈化柱處理所得的空白綠茶基質和1%乙酸-乙腈提取溶劑配制濃度為50 μg/L的14種混合農藥標準溶液,重復測定3次,考察14種農藥在綠茶中的基質效應。由表3可以看出,綠茶基質對14種農藥的基質效應不同,10種農藥ME>105%,為基質增強;2種農藥ME<85%,為基質抑制;2種農藥ME在85%~105%之間,表示無基質效應。因此,對茶葉農殘檢測,采用空白基質配制標準曲線進行定量分析以減弱基質效應的影響,提高定量結果的準確性。

表3 14種農藥的線性方程、決定系數、檢出限、定量限、基質效應和最大殘留限量Table 3 Linear equations, coefficient of determination (R2), LODs, LOQs, matrix effects and the maximum residue limits of 14 pesticides
2.5 線性關系、檢出限、定量限
由表3可知,14種農藥在相應的線性范圍內線性關系良好,R2>0.99。以信噪比(S/N)為3和10時對應的空白樣品添加濃度為檢出限(Limit of detection,LOD)和定量限(Limit of quantification, LOQ),14種農藥LOD在0.001~2.000 μg/L之間,LOQ為0.003~5.000 μg/L。根據GB 2763-2021《食品安全國家標準 食品中農藥最大殘留限量》,除涕滅威在茶葉中的最大殘留量未做規定,其余13種農藥的檢出限和定量限均遠低于規定的最大殘留量,說明本文方法滿足實際檢測需求。
2.6 回收率和精密度
如表4所示,在向空白綠茶和紅茶樣品中添加低、中、高濃度(100、500、800 μg/kg)三個濃度水平下,方法的平均回收率為71.2%~117.4%,RSD為1.1%~14.2%,說明該方法對綠茶和紅茶中14種農藥殘留檢測的準確度和精密度良好,能夠滿足茶葉樣品多農藥殘留分析的要求。

表4 14種農藥在綠茶和紅茶中的加標回收率和相對標準偏差(n=6)Table 4 Recoveries and RSDs of the 14 pesticides spiked in green tea and black tea (n=6)
2.7 實際樣品檢測
利用本文方法對市售的30種茶葉進行檢測,如表5所示,有6種茶葉檢出目標農藥,其中5種茶葉檢出啶蟲脒,殘留量為2.4~14.1 μg/kg;2種茶葉檢出克百威,殘留量分別為1.9、23.0 μg/kg;2種茶葉檢出吡蟲啉,殘留量分別為1.9、33.7 μg/kg;1種茶葉檢出茚蟲威,殘留量為3.0 μg/kg;1種茶葉檢出噻蟲嗪,殘留量為2.4 μg/kg。上述茶葉中農藥殘留均未超過國家標準GB 2763-2021規定的最大殘留限量值。

表5 6種檢出目標農藥的茶葉中農藥殘留濃度(μg/kg)Table 5 Concentration of targeted pesticides detected in six tea samples
3 結論
本文采用QuEChERS前處理方法結合UPLCMS/MS技術,建立了茶葉中啶蟲脒、涕滅威、克百威、內吸磷、滅線磷、吡蟲啉、茚蟲威、甲胺磷、滅多威、氧化樂果、甲基對硫磷、硫環磷、丙溴磷、噻蟲嗪等14種農藥殘留的快速檢測方法。通過優化提取液組分、QuEChERS凈化柱種類等,14種農藥的LOQ可達到0.003~5.000 μg/L,在3個添加水平下的回收率為71.2%~117.4%,能夠滿足茶葉農藥殘留限量指標的快速檢測要求。對30種市售茶葉樣品進行檢測,其中6種茶葉檢出目標農藥,測得農藥殘留濃度均未超過國家限量標準。本文方法具有操作簡便快捷、凈化效果好、靈敏度和準確率高等優點,為高效和準確地評價茶葉的質量安全提供了新的參考。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
