柱前衍生化反相高效液相色譜法測定春雷霉素
2022-06-16 16:27:14張佳慶楊聞翰侯德粉
現代農藥 2022年3期
張佳慶,楊聞翰,張 丹,侯德粉
(沈陽沈化院測試技術有限公司,沈陽 110021)
春雷霉素(Kasugamycin)是一種低毒、內吸性殺菌劑,通過內吸治療作用來發揮其藥效。春雷霉素的前身是一種由小金色放線菌產生的抗菌素[1],最早由中國科學院微生物研究所于1964年在江西土良地區發現。自春雷霉素上市以來,在全世界范圍內被廣泛應用于農業生產上[2]。它對稻瘟病、桃樹流膠病、穿孔病、西瓜細菌性角斑病等病害的防治具有顯著作用[3]。
春雷霉素屬于堿性物質,其結構特點為極性大,易溶于水,故在液相色譜行為上表現為色譜的保留時間短[4-5],對于結構相近的生產雜質,很難達到理想的分離效果,從而給檢測的準確度造成了影響。目前,對于春雷霉素含量測定的液相色譜分析方法,一般選擇較為復雜的流動相配制體系,通常采用一定濃度的十二烷基磺酸鈉水溶液作為基本的流動相,再配合一定比例的有機相。然而,大濃度的緩沖鹽體系對色譜柱損耗嚴重,影響色譜柱的壽命,提高了檢測成本。本研究開發了柱前衍生方法,對春雷霉素的結構進行了衍生化,通過增加衍生基團,使春雷霉素具有良好的紫外吸收特性。通過調整春雷霉素的極性,可實現在乙腈-水為流動相體系下,對春雷霉素的原藥含量進行分析。此法檢測靈敏度高,重現性好,經濟實用,并且可以同時實現對春雷霉素原藥中一般雜質的有效分離,為春雷霉素產品的分析提供了一種創新性的思路。
1 材料與方法
1.1 儀器與試劑
Acquity-Micromass Quattro premier液質聯用儀,美國沃特世公司;Dionex U3000高效液相色譜儀,美國賽默飛世爾公司;Agilent Eclipse XDB-C18色譜柱(4.6 mm×150 mm,5 μm),安捷倫科技(中國)有限公司;XS205DU電子天平,梅特勒托利多科技(中國)有限公司;四硼酸鈉、甲醇、乙腈,以上均為色譜純,美國SIGMA ALDRICH公司;醋酸(分析純),國藥集團化學試劑有限公司;標準品春雷霉素(純度98.0%),國家農藥質檢中心(沈陽);春雷霉素原藥,河北安米諾氨基酸科技股份有限公司;氯甲酸-9-芴甲酯(FMOC-Cl),北京百靈威科技有限公司。
1.2 儀器條件
色譜條件。柱溫:30℃;流速:1.0 mL/min;進樣量:5 μL;檢測器波長:265 nm。采用梯度洗脫,洗脫程序見表1。

表1 流動相梯度比例
質譜條件。離子源模式:ESI+;電離電壓:3.5 kV;錐孔電壓40 V;氣簾氣流速:50 L/h;裂解氣流速:600 L/h;裂解氣溫度:400℃;離子源溫度:120℃;分子量采集范圍設置:100~600(m/z)。
1.3 溶液配制
1.3.1 標準溶液的配制
稱取春雷霉素標準品0.05 g(精確至0.000 1 g)置于燒杯中,用水溶解,稀釋并定容于50 mL容量瓶中作為標樣母液。取1.0 mL母液置于燒杯中,再加入1 mL 125 mmol/L四硼酸鈉溶液來調節酸堿度,之后加入1.5 mL 4 g/L FMOC-Cl乙腈溶液作為衍生化試劑。將溶液放置在30℃水浴中反應20 min,待衍生完全后,將溶液冷卻至室溫,再用甲醇-水(1∶2,V/V)稀釋并定容于10 mL容量瓶中,用0.45 μm濾膜過濾,備用。
1.3.2 試樣溶液的配制
稱取春雷霉素樣品0.05 g(精確至0.000 1 g)置于燒杯中,用水溶解,稀釋并定容于50 mL容量瓶中作為樣品母液。其余操作步驟與標準溶液的配制相同。
1.4 測定
待儀器狀態穩定后,對標樣溶液和試樣溶液進行測定,取樣品和標樣2針的平均峰面積進行公式的計算。
1.5 數據分析
根據標樣溶液和試樣溶液的峰面積,按式(1)計算樣品中春雷霉素的質量分數w。
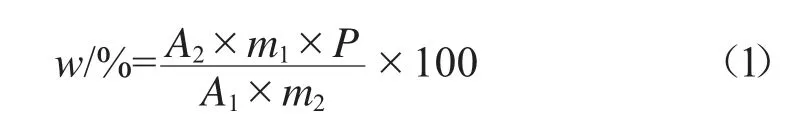
式中:A1、A2分別為標樣溶液和試樣溶液中春雷霉素衍生物峰面積的平均值,mAU·min;m1、m2分別為標樣和試樣質量,g;P為標樣中春雷霉素的質量分數,%。
2 結果與分析
2.1 檢測波長的選擇
對春雷霉素標樣及其衍生物進行紫外光譜掃描(圖1)。春雷霉素未衍生化之前的最大紫外吸收峰在207 nm附近,且無其他波長的特征吸收峰,所以在對未衍生化的春雷霉素進行色譜分析時,只能選擇較低的紫外波長,此段波長的干擾較大,而經過衍生化的春雷霉素在265 nm附近出現了特征的紫外吸收峰。在該波長處,樣品的靈敏度較高,無低波數的溶劑干擾,能夠滿足液相色譜紫外檢測器的分析要求,因此選擇的檢測波長為265 nm。
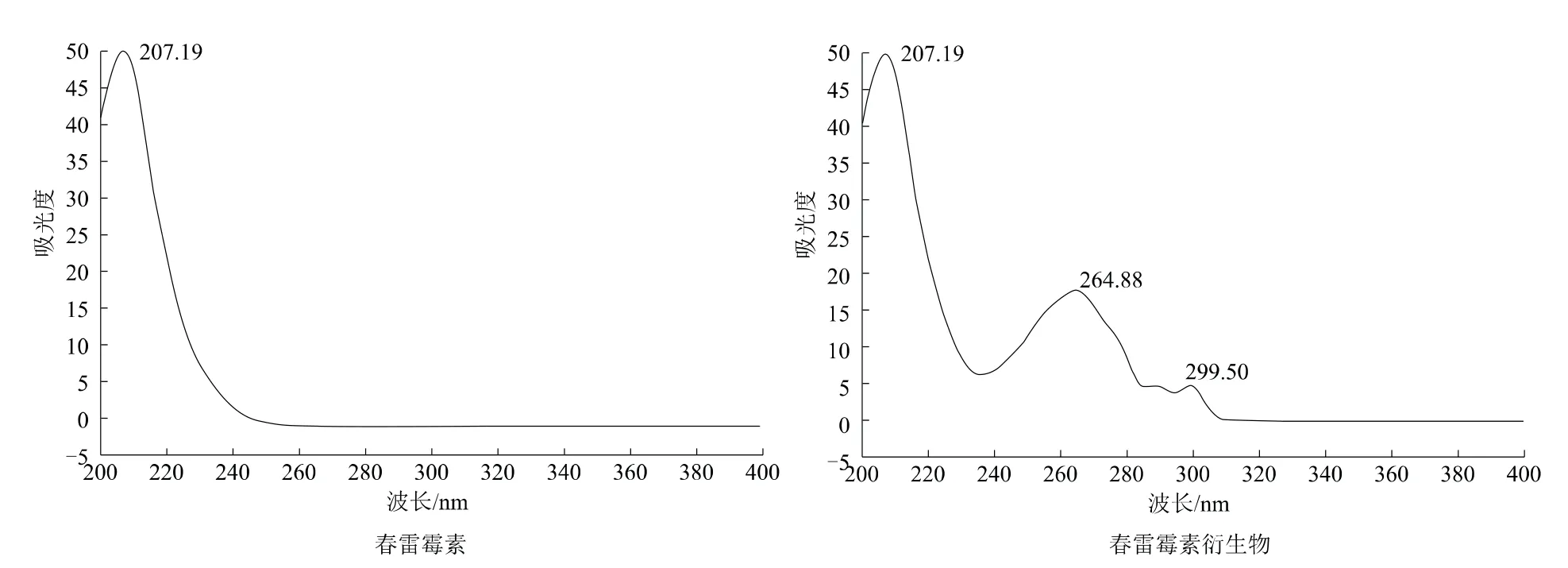
圖1 春雷霉素及其衍生物的紫外吸收對比圖
2.2 春雷霉素衍生物的色譜分析結果
經過衍生反應(圖2),向春雷霉素引入衍生化試劑氯甲酸-9-芴甲酯(FMOC-Cl)中的熒光團,從而實現高效液相色譜分離及測定。同時,過量的FMOC-Cl會完全水解成衍生副產物FMOC-OH,其與春雷霉素衍生物的結構相似,因此具有相同的熒光激發和檢測波長。在以上色譜條件下,春雷霉素衍生物的保留時間為6.8 min;其水解衍生副產物FMOC-OH的保留時間為12.1 min(圖3)。
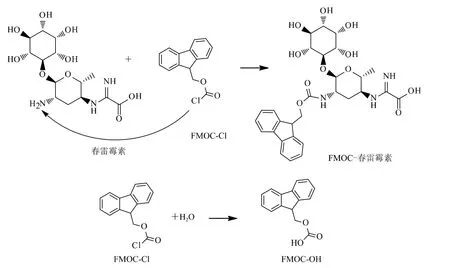
圖2 春雷霉素柱前衍生化反應示意圖
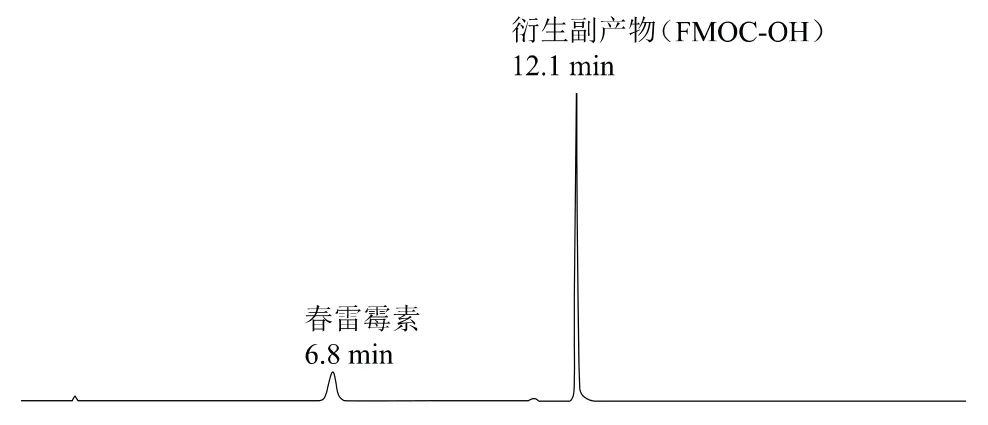
圖3 春雷霉素衍生后的液相色譜圖
2.3 春雷霉素衍生物的質譜分析結果
將春雷霉素標樣溶液在質譜條件下進行試驗,通過質譜的斷裂機理確定衍生化試劑(FMOC-Cl)對春雷霉素的衍生位置,以及春雷霉素衍生物的結構。春雷霉素衍生物的質譜圖,見圖4。
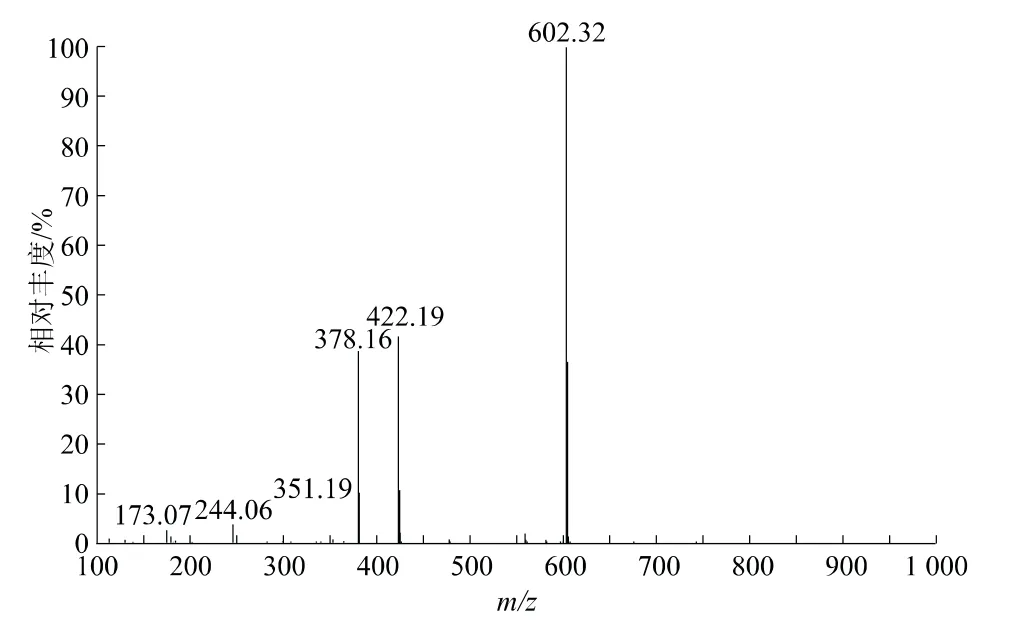
圖4 春雷霉素衍生物質譜圖
2.4 線性范圍
按照系列稀釋方法配制6個質量濃度的春雷霉素標準品溶液,在1.2所述色譜條件下進行測定。以質量濃度和峰面積為橫、縱坐標繪制標準曲線,來驗證方法的線性關系。線性方程為y=4 887.3x-29.319,相關系數(R2)為0.999 5(圖4),R2>0.999,則春雷霉素衍生物的色譜峰面積與其質量濃度在0.039 6~0.415 4 mg/L范圍內線性關系良好,
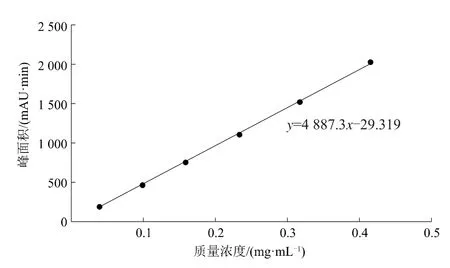
圖5 春雷霉素衍生物標準曲線
2.5 精密度與回收率的測定
按試樣溶液的制備方法配制6個春雷霉素溶液。根據以上操作條件測定這6個樣品溶液中春雷霉素的含量,結果見表2。在此春雷霉素樣品中選取6個標樣的添加水平,按本方法測定其中的春雷霉素含量,并計算添加回收率,結果見表3。從表2中可以看出,所配制的6個春雷霉素樣品中相對標準偏差(RSD)為0.4%;從表3中可以看出,計算得到的回收率為98.56%~99.31%。
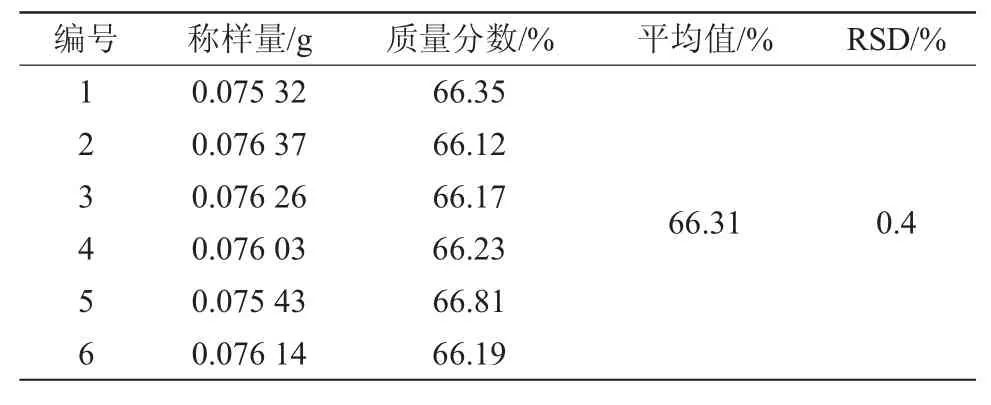
表2 春雷霉素精密度試驗結果
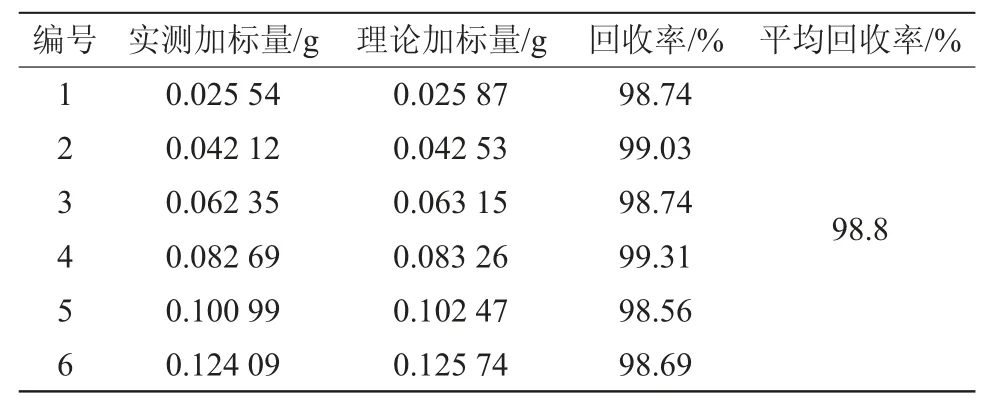
表3 春雷霉素回收率試驗結果
2.6 春雷霉素衍生物結構的確證
根據春雷霉素衍生物的質譜圖(圖4),m/z 602、422、351和378為主要的質譜峰,通過質譜的一般斷裂機理來確證春雷霉素衍生物的具體結構(圖6),以此驗證春雷霉素衍生物的分子量和各官能團信息。
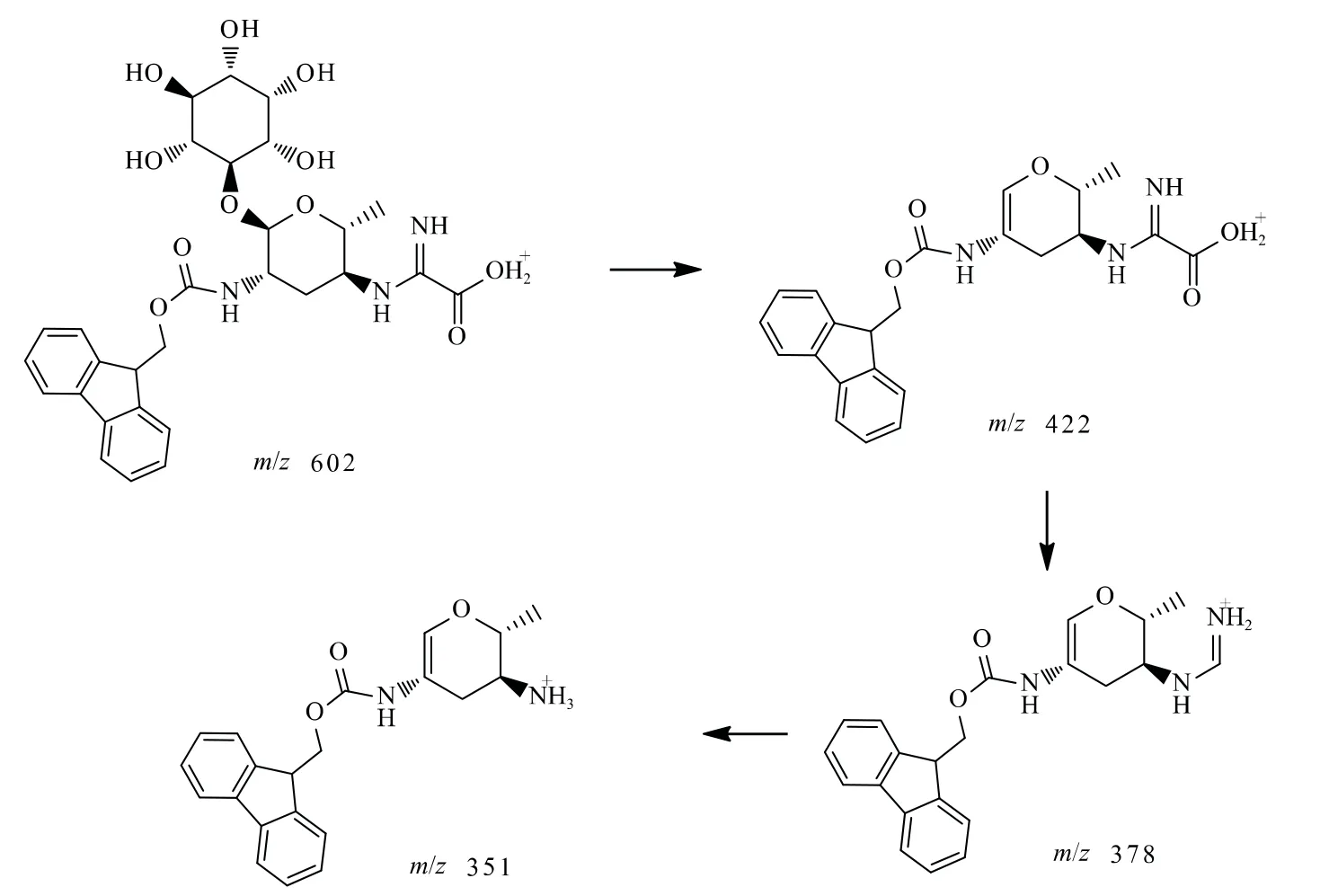
圖6 春雷霉素衍生物質譜斷裂機理示意圖
3 結論
本文創新性地建立了柱前衍生方法對春雷霉素進行液相色譜分析,使用乙腈-水的流動相體系,解決了在傳統的春雷霉素分析方法中需要使用緩沖鹽溶液作為流動相而導致的體系平衡緩慢、靈敏度低、對色譜柱損耗大以及雜質分離不理想等問題,提高了對春雷霉素產品分析的簡便性及可靠性。此外,本文也通過液質聯用方法對春雷霉素衍生物進行了結構分析,闡明了春雷霉素柱前衍生化反應原理,并通過春雷霉素衍生化前后紫外全波長掃描圖的對比,進一步證明了方法的科學性。本方法具有較高的精密度與準確度,可快速分離及測定春雷霉素有效成分的含量,線性相關性好,可為春雷霉素產品的分析提供新的思路,適用于各企業及相關檢測機構對春雷霉素產品質量的監督與把控。
