HPLC 法測定頭孢氨芐中L- 頭孢氨芐雜質的研究
2022-06-17 07:46:40王新輝
煤炭與化工 2022年5期
關鍵詞:方法
王新輝
(華北制藥股份有限公司倍達分廠,河北 石家莊 052165)
0 引言
頭孢氨芐(Cefalexin) 為廣譜類抗生素,可以抑制細胞壁的合成,使細胞內容物膨脹至破裂溶解,殺死細菌。為確保仿制藥與原研藥品質量和療效一致,保證公眾用藥安全,中國開始推行一致性評價工作,要求仿制藥廠家深入開展對藥品的雜質及質量的研究工作。在《中華人民共和國藥典》2020 版中頭孢氨芐采用梯度測定有關物質,能有效分離7- 氨基去乙酰氧基頭孢烷酸(雜質B 即7-ADCA) 和α- 苯甘氨酸(雜質A) 等雜質,但L- 頭孢氨芐不能基線分離。故通過對流動相、梯度洗脫方法等進行優化,建立檢測頭孢氨芐中L-頭孢氨芐的方法,并對該方法進行驗證。
1 儀器和材料
1.1 儀器
高效液相色譜儀。
電子天平。
pH 計。
1.2 材料
L- 頭孢氨芐對照品,批號為2618-043A2。
頭孢氨芐對照品,批號為1304008-201411。
氫氧化鈉,AR 批號為20210107。
無水磷酸二氫鈉,AR 批號為P1533702。
甲醇,HPLC 批號為21105212。
2 方法優化過程
經查閱各國藥典,對比頭孢氨芐有關物質所使用的方法,各國藥典有關物質檢查方法所用色譜柱的種類相同,均為C18 柱;中國藥典和歐洲藥典(英國藥典) 所用流動相緩沖鹽的pH 值、洗脫程序和檢測波長相同,但緩沖鹽的種類和濃度不同;與美國藥典所用流動相、洗脫程序和檢測波長均不同。對各國藥典的有關物質檢查方法進行對比優化,確定最佳檢測方法。
2.1 方法選擇
按照中國藥典、歐洲藥典(英國藥典) 和美國藥典的有關物質檢查方法,分別配制供試品溶液和供試品溶液高溫破壞溶液,分別進樣,記錄色譜圖。美國藥典方法的靈敏度低,不采用,中國藥典和歐洲藥典高溫破壞溶液色譜圖如圖1 所示。

圖1 中國藥典和歐洲藥典高溫破壞溶液色譜圖Fig.1 Chromatograms for high-temperature destruction solutions using Ch.P and EP method
由圖1 可看出,中國藥典方法主峰的分離度要優于歐洲藥典方法,故在中國藥典頭孢氨芐有關物質的方法上進行優化對L- 頭孢氨芐進行檢驗。
2.2 流速選擇
在相同色譜柱、流動相和柱溫條件下,分別設定流速為1.0 mL/min、1.2 mL/min 和1.5 mL/min,分別精密量取供試品高溫破壞溶液各20 μL 注入液相色譜儀,記錄色譜圖。比較不同流速下雜質的分離情況,不同流速下色譜圖如圖2 所示。

圖2 不同流速下色譜圖Fig.2 Chromatograms for Different flowvelocity
由圖2 可看出,流速1.5 mL/min 下主峰與相鄰雜質峰的分離度明顯優于其他2 個流速,故方法選擇流速為1.5 mL/min。
2.3 緩沖鹽濃度選擇
在相同色譜柱、流速和柱溫條件下,分別配制0.05 mol/L、0.1 mol/L 和0.2 mol/L 的磷酸二氫鈉緩沖溶液(pH 值為5.0) 作為流動相A,比較系統適用性溶液的分離情況,不同濃度流動相A 系統適用性溶液色譜圖對比如圖3 所示。

圖3 不同濃度流動相A系統適用性溶液色譜圖對比Fig.3 Comparison of system suitability chromatograms for different concentrations of mobile phase A
由圖3 可看出,緩沖鹽濃度為0.05 mol/L 時系統適用性溶液中主峰與后臨雜質峰間不能達到分離要求,濃度為0.1 mol/L 和0.2 mol/L 時,主峰與相鄰雜質峰之間均能達到分離要求。故確定0.1 mol/L磷酸二氫鈉溶液(pH 值為5.0) 作為有關物質檢測流動相A。
3 方法確定
3.1 色譜條件
色譜柱:CAPCELL PAK C18 MGⅡ(4.6*150 mm,5μm),以0.1 mol/L 磷酸二氫鈉(pH 5.0)為流動相A,以甲醇為流動相B,流速為1.5 mL/min,進樣量為20μL,檢測器波長為220 nm。梯度洗脫程序為:

B/%梯度洗脫時間/min 0153742555660 A/%989370703098982730307022
3.2 溶液的制備
3.2.1 流動相A
流動相A 0.1 mol/L 磷酸二氫鈉(pH 值為5.0)溶液。以流動相A 作為空白溶液。
3.2.2 L- 頭孢氨芐對照溶液
L- 頭孢氨芐對照溶液精密稱取L- 頭孢氨芐對照品約2.5 mg,置于5 mL 容量瓶中,使用流動相A 稀釋并定容。移取上述溶液0.02 mL,置于5 mL容量瓶中,加流動相A 稀釋并定容。
3.2.3 混合定位溶液
混合定位溶液取EP 雜質A(α- 苯甘氨酸)對照品和雜質B(7-ADCA) 對照品各5 mg,分別置兩個10 mL 容量瓶中,加流動相稀釋并定容。稱取頭孢氨芐對照品5 mg,置于5 mL 容量瓶中,用流動相稀釋并定容。取上述各對照品溶液各1.0 mL,置于同一50 mL 容量瓶中,加流動相A 稀釋并定容,作為有關物質混合定位溶液。
3.2.4 雜質混合定位溶液
雜質混合定位溶液精密稱取L- 頭孢氨芐對照品約2.5 mg,置于5 mL 容量瓶中,使用流動相A稀釋并定容。移取上述溶液0.02 mL,置于5 mL 容量瓶中,加流動相A 稀釋并定容。移取上述溶液0.1 mL 與“有關物質混合定位溶液”0.1 mL 混合,作為雜質混合定位溶液。
3.2.5 供試品溶液
供試品溶液取頭孢氨芐供試品10 mg,置100 mL 容量瓶中,加流動相A 稀釋并定容。
3.2.6 對照品溶液對照品溶液
精密量取供試品溶液1.0 mL 置于100 mL 容量瓶中,加流動相A 稀釋并定容。
3.2.7 供試品溶液高溫破壞
供試品溶液高溫破壞取供試品溶液適量,在80 ℃水浴中加熱60 min。
3.3 測定方法
測定方法按方法描述分別稱取L- 頭孢氨芐對照品溶液、對照品溶液和供試品溶液,并分別注入液相色譜儀,記錄色譜圖。按外標法以峰面積計算,含L- 頭孢氨芐雜質的量不得過0.2%。
4 方法驗證
4.1 專屬性考察
取雜質混合定位溶液和流動相A(空白溶液)各10 μL,注入高效液相色譜儀,空白溶液在各目標峰處無明顯干擾。雜質混合定位溶液中各目標峰的保留時間與加標供試品溶液中的保留時間一致,主峰與相鄰雜質分離度分別為2.27、3.26。均滿足專屬性要求。空白溶液色譜圖如圖4 所示。

圖4 空白溶液色譜圖Fig.4 Chromatograms of blank solution
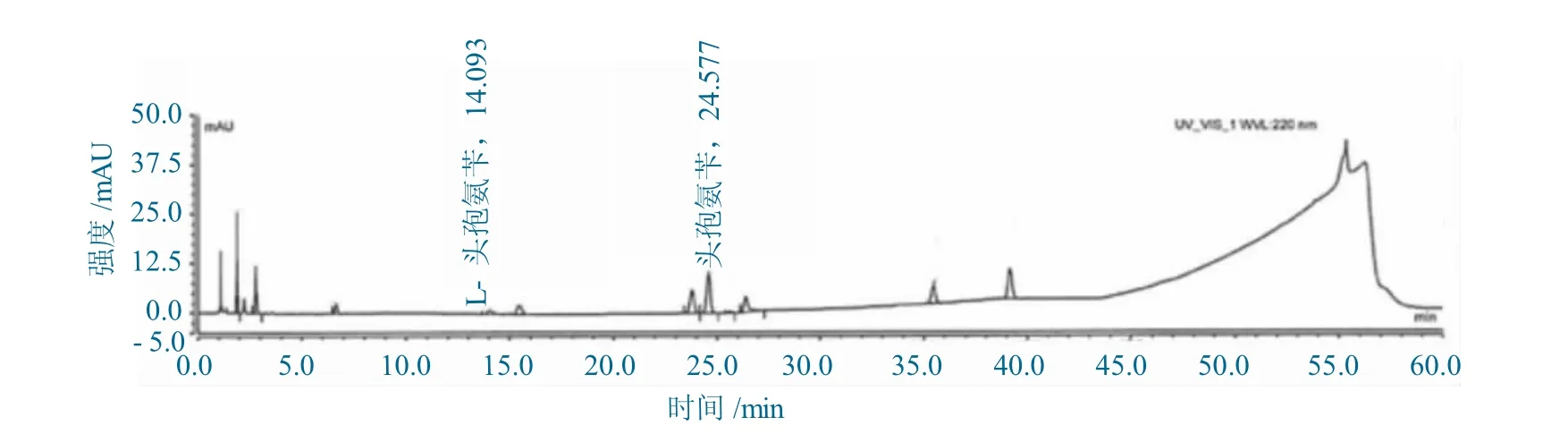
圖5 L- 頭孢氨芐定位圖Fig.5 Chromatograms of Location map of L-Cefalexin
4.2 定量限/檢測限考察
儲備液:取L- 頭孢氨芐對照品2.5 mg,置于5 mL 容量瓶中,用流動相A 稀釋并定容。轉移上述溶液0.4 mL,置2 mL 容量瓶中,用流動相A 稀釋并定容。取頭孢氨芐對照品2.5 mg,置于5 mL容量瓶中,用流動相A 稀釋并定容,轉移上述溶液0.4 mL,置2 mL 容量瓶中,用流動相A 稀釋并定容。取上述兩溶液各0.3 mL,置同一10 mL 容量瓶中,用流動相A 稀釋并定容。
定量限溶液:取上述儲備液1.0 mL,置10 mL容量瓶中,用流動相A 稀釋并定容。
檢測限溶液:取定量限溶液2.5 mL,置5 mL容量瓶中,用流動相A 稀釋并定容。
檢測結果:L- 頭孢氨芐信噪比S/N 為17.0 ~22.1,滿足信噪比≥10∶1 的要求,定量限濃度為0.3180 μg/mL,相當于供試品的濃度百分比0.03%,連續進樣6 針的RSD 為5.8%。
L- 頭孢氨芐信噪比S/N 為7.5,滿足信噪比≥3∶1 的要求,檢測限濃度為0.1590 μg/mL,相當于供試品的濃度百分比0.02%,均滿足要求。
4.3 線性范圍
取定量限溶液0.4 mL,置于20 mL 容量瓶中,用流動相A 稀釋并定容。作為線性儲備溶液。
分別取線性儲備溶液0.5,1.0,1.5 和2.0 mL轉移至10 mL 容量瓶中,并用流動相A 稀釋定容。將定量限溶液及上述4 種溶液分別進高效液相色譜儀,記錄色譜圖。L- 頭孢氨芐線性方程為:y=0.3022x- 0.0078,相關系數r 為0.9997。L- 頭孢氨芐濃度范圍:0.3180 ~2.1199 μg/mL,相當于供試品的濃度百分比0.03%~0.21%。
4.4 準確度
準確度儲備液:取定量限溶液0.4 mL,置于20 mL 容量瓶中,用流動相A 稀釋并定容。
精密稱取頭孢氨芐供試品10 mg 9 份置于10 mL 容量瓶中,分為以下3 組:
(1) 分別移取1.0 mL 定量限儲備液于容量瓶中,并用流動相A 稀釋定容。
(2) 分別移取1.0 mL 準確度儲備液于容量瓶中,并用流動相A 稀釋定容。
(3) 分別移取1.5 mL 準確度儲備液于容量瓶中,并用流動相A 稀釋定容。
檢驗結果說明,LOQ 濃度水平加標供試品平均回收率為93%~117%(n=3),回收率的RSD 為14%(n=3),其余水平加標供試品溶液各水平的平均回收率均為113%~119%(n=3),回收率的RSD為2%~4%(n=3),符合要求。
4.5 重復性
精密稱取頭孢氨芐供試品10 mg 6 份置于10 mL 容量瓶中,并各轉移1 mL 準確度儲備液于該容量瓶中,分別進樣,計算6 份樣品中L- 頭孢氨芐含量的RSD 值,為2%,符合重復性的要求。
5 結語
本文參考各國藥典中頭孢氨芐有關物質的測定方法,通過一系列的試驗研究及對方法的優化,建立了梯度的方法對L- 頭孢氨芐的雜質進行檢測,并通過對方法的專屬性、檢測限、定量限、準確度等進行驗證,結果表明,該方法靈敏度好、專屬性強、準確度高,能有效分析頭孢氨芐及其雜質L-頭孢氨芐,為更好的控制頭孢氨芐中的雜質提供了依據,可用于頭孢氨芐中L- 頭孢氨芐的檢測和對頭孢氨芐的質量控制。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
