
多粘類芽孢桿菌LY1 的全基因組測序及分析
2022-06-22 08:26:06聶俊輝張立新郭建軍
湖南農業科學 2022年5期
聶俊輝,王 平,張立新,王 通,郭建軍,曾 靜,袁 林
(1.江西省科學院微生物研究所,江西 南昌 330096;2.上饒市農林水科學研究中心,江西 上饒 334000)
多粘類芽孢桿菌(Paenibacillus polymyxa)屬芽孢桿菌科(Bacillaceae)類芽孢桿菌屬(Paenibacillus),是一種產芽孢的革蘭氏陽性菌[1]。多粘類芽孢桿菌在自然界中分布廣泛,在玉米、高粱、小麥等多種植物體內外以及根際土壤中均有被發現,是一類重要的根際有益菌[2]。多粘類芽孢桿菌對植物生長發育的有益作用主要表現在促進植物產生激素、促進植物吸收養分、促進植物光合作用、增強植物固氮作用等方面[3];同時,多粘類芽孢桿菌還能通過分泌抗菌殺蟲物質如抗菌肽、細菌素、脂肽等增強植物抵御病蟲害的能力,是一種不可多得的生物防治原料,目前已廣泛應用于農業生產領域,具有較高的經濟價值[4-5]。
近年來,高通量測序技術快速發展,使得細菌基因組的高效解析成為現實,為細菌的功能及其作用機制研究提供了重要手段[6-7]。趙興麗等[8]通過高通量測序技術發現土壤中有益微生物的含量在一定程度上可間接反映茶樹的抗病性,有益微生物菌群所占比率與發病程度呈負相關。陳德純等[9]對一株牦牛源產細菌素植物乳桿菌的基因組進行測序,發現其基因組中包括多個細菌素基因以及細菌素轉運系統的功能序列,為該菌株的應用研究提供了重要的參考數據。高通量測序技術的普及有效地促進了微生物的研究。
目前,有關多粘類芽孢桿菌的研究多集中在其生物特性及其對植物與生物防治的作用機理上,而對其基因序列的分析研究報道較少[10-12]。筆者從土壤中分離出一株多粘類芽孢桿菌LY1,利用基因組測序技術全面解析LY1 菌株的基因組序列,再通過生物信息學的方法比較不同菌株基因間的差異性和相似性,探索其基因結構特征,明確其基因功能,從全基因組水平了解其系統發育進化關系,以期為全面了解多粘類芽孢桿菌的遺傳背景,實現多粘類芽孢桿菌的遺傳改造提供基本線索。
1 材料與方法
1.1 供試菌株
多粘類芽孢桿菌LY1 從土壤中分離,由江西省科學院微生物研究所分離保存。
1.2 試驗方法
1.2.1 測序及數據質控取菌體委托上海生工生物工程股份有限公司使用Illumina Hiseq platform 進行基因組測序。Illumina Hiseq?得到的原始圖像數據文件經CASAVA 堿基識別 (Base Calling)分析轉化為原始測序序列(Sequenced Reads),對原始數據質量值等信息進行統計,并使用FastQC 對樣本的測序數據質量進行可視化評估。測序得到的原始數據,里面含有帶接頭的、低質量的序列。為了保證信息分析質量,必須對原始數據進行過濾,得到Clean 數據。隨機從Clean 數據中抽取10 000 條序列與NCBI NT 數據庫進行blastn 比對,取evalue ≤1E-10 并且相似度>90%、coverage >80%的比對結果,計算其物種分布,同時進行污染檢測。
1.2.2 數據拼裝使用SPAdes 對二代測序數據進行拼裝。SPAdes 首先會對原始序列進行序列錯誤校正,然后通過多Kmer 值進行組裝,最終綜合各Kmer 值組裝結果獲得最佳結果。再采用GapFiller 對拼接得到的contig 進行Gap 修補,最后采用PrInSeS-G 進行序列矯正,修正拼接過程中的剪輯錯誤以及小片段的插入缺失。
1.2.3 基因預測與注釋采用Prokka 對組裝結果進行基因元件預測。Prokka 是一系列基因元件預測工具的集合,調用Prodigal 預測編碼基因,Aragorn 預測tRNA,RNAmmer 預測rRNA,Infernal 預測miscRNA,預測出的各類基因元件匯總并完成初步注釋。采用Repeat Modeler 對組裝結果進行重復序列Denovo 預測,再利用RepeatMasker 尋找基因組區段上各類型重復序列出現的位置和頻率。NCBI Blast+用于CDD、KOG、COG、NR、NT、PFAM、Swissprot、TrEMBL注釋。基于Swissprot 和TrEMBL 的蛋白注釋結果根據Uniprot 的注釋信息得到GO 注釋。使用KAAS,KEGG Automatic Annotation Server 進行KEGG 注釋。
1.2.4 系統發育樹構建將基因預測得到的16S rRNA序列與NCBI 的16S 數據庫進行Blast 比對,設置參數identify >95。然后選取identify 最高的前30 條16S rRNA序列,利用muscle軟件進行序列多重比對后,采用FastTree 軟件構建系統發育樹。
2 結果與分析
2.1 測序數據質控
二代測序和三代測序原始數據經過去除帶接頭的、低質量的序列,過濾后得到Clean 數據。Illumina Hiseq 原始測序數據過濾處理后,總共得到6 495 188個Clean Reads,總堿基數為951 905 276 bp,平均Read長度為146.56 bp;其中,Q10、Q20、Q30 的比例分別為100.00%(堿基數951 903 300 bp)、98.18%(堿基數934 538 164 bp)、93.79%(堿基數892 808 488 bp);GC含量為45.63%(堿基數434 383 207 bp)。PacBio RSII原始測序數據過濾處理后,總共得到333 829 個Clean Reads,總堿基計數為925 726 342 bp,平均Reads 長度2 773.06 bp;其中,Reads ≥1 000 bp 、≥5 000 bp和≥10 000 bp 的分別占比47.34%、11.45%和5.30%,而Bases in Reads ≥1 000 bp、≥5 000 bp 和≥10 000 bp 的分別占比90.25%、61.52%和46.25%;GC 含量為44.40%。綜上所述,2 組數據質量較高,可用于下一步基因組組裝。
2.2 基因預測與基因組基本特征
2.2.1 基因組的組成與信息2 組測序數據經過序列矯正、Gap 修補、剪輯錯誤修復、拼裝等程序后獲得菌株的基因組信息,如圖1 所示,多粘類芽孢桿菌LY1 的基因組為一條環狀閉合DNA,大小 5 765 474 bp,共預測到6 086 個蛋白質編碼基因、162 個 tRNA基因、42 個rRNA 基因、74 個small RNA ;最短基因長度為70 bp,最長基因長度為42 252 bp,基因平均長度為889.05 bp,長度≥500 bp 的基因有4 107 個,長度≥1 000 bp 的基因有1 916 個;重復區域計數為216 個,重復比例為1.45%;Low_complexity 為25 個,Simple_repeat 為117 個。基因長度和GC 含量的分布如圖2 所示。

圖1 多粘類芽孢桿菌LY1 的基因組圖
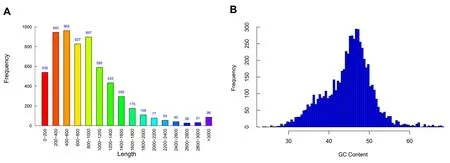
圖2 多粘類芽孢桿菌LY1 的基因長度(A)和GC 含量分布(B)
2.2.2 基因組注釋將預測的編碼基因與多個數據庫進行比對,注釋到CDD(Conserved Domain Database)、KOG(euKaryotic Ortholog Groups)、NR(NCBI nonredundant protein sequences)、PFAM(Protein family)、Swissprot(A manually annotated and reviewed protein sequence database)、TrEMBL、GO(Gene Ontology)、KEGG(Kyoto Encyclopedia of Genes and Genomes)的基因比例分別為72.92%、64.27%、98.14%、69.57%、66.32%、97.52%、66.66%和37.92%,unigenes 總共5 813 個,同時注釋到NR、KEGG、Swissprot、COG的基因有2 133 個,注釋到所有數據庫的基因共有2 066 個,占unigenes 的35.54%。
2.2.3 NR 數據庫注釋由上面的數據可知,注釋到NR 數據庫的基因最為豐富,共有5 705 個,占比98.14%。NR 數據庫是一個非冗余的蛋白質數據庫,內容全面,通過與NR 數據庫的對比,可以查看物種轉錄本序列與相近物種的近似情況以及同源序列的功能信息[13]。由圖3 可知,鑒定到LY1 菌株有3 792 個基因與多粘類芽孢桿菌同源,與Paenibacillus polymyxaM1 菌株的相似性相對較高,在M1 的編碼基因中注釋到318 個基因,而在Paenibacillus polymyxaSQR-21、Paenibacillus polymyxaSC2 編 碼 基因中則分別注釋到76 和29 個基因。
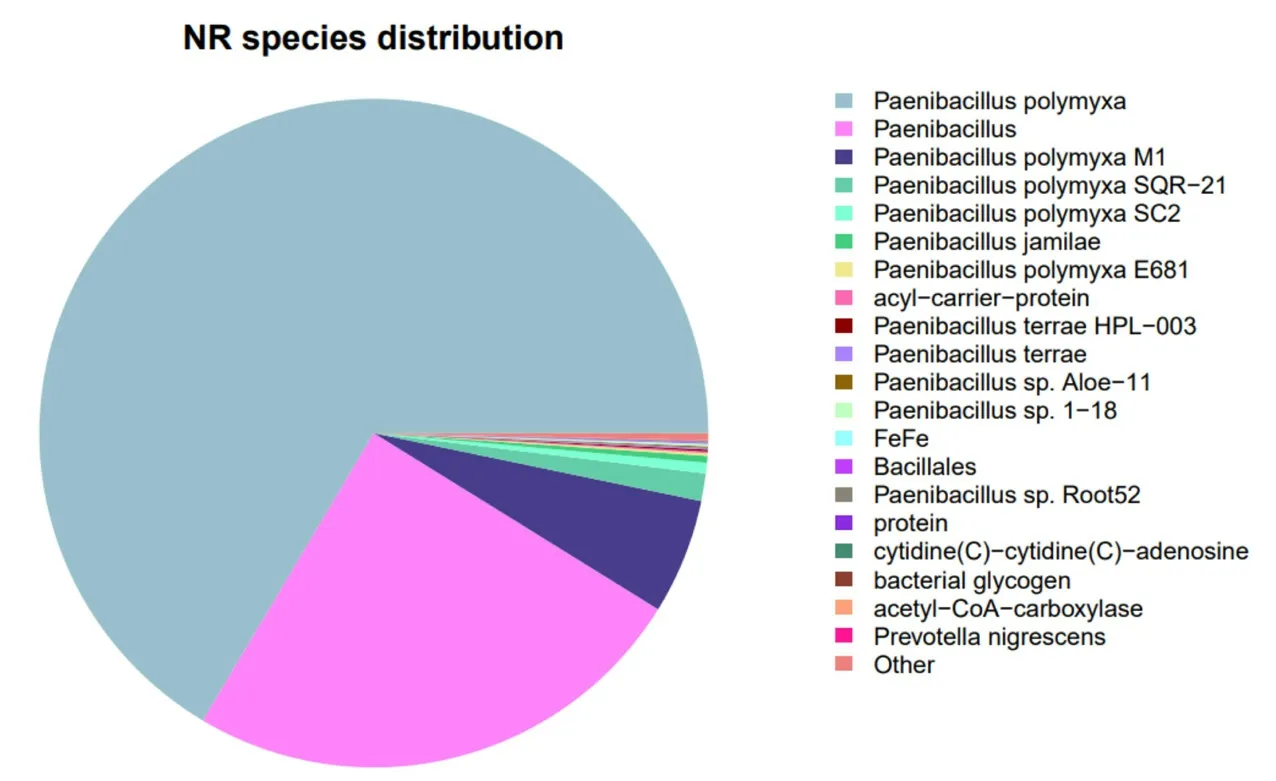
圖3 多粘類芽孢桿菌LY1 基因的NR 數據庫比對
2.3 基因功能注釋
2.3.1 GO 功能注釋GO (Gene Ontology) 是一個國際標準化的基因功能分類體系,提供了一套動態更新的標準詞匯表(controlled vocabulary)來全面描述生物體中基因和基因產物的屬性[14]。GO 總共有3 個ontology,分別描述基因的分子功能(MF,molecular function)、細胞組分(CC,cellular component)、參與的生物過程(BP,biological process)[15]。通過將基因進行GO 注釋和分類,可判斷基因參與的主要功能。多粘類芽孢桿菌LY1 基因的3 大分類統計結果如圖4 所示。在生物過程中,分類到代謝過程(metabolic process)和細胞過程(cellular process)中的基因占比最多,分別為2 370(40.77%)和2 266 個(38.98%);在細胞組分分類中,分類到細胞(cell)和細胞組成(cell part)中的基因占比最多,均為2 045 個(35.18%);在分子功能分類中,分類到催化活性(catalytic activity)和結合(binding)中的基因占比最多,分別為2 345(40.34%)和2 080 個(35.78%)。
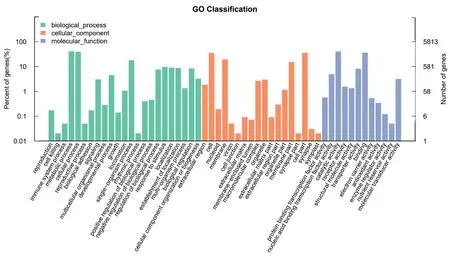
圖4 多粘類芽孢桿菌LY1 的GO 功能注釋
2.3.2 KEGG 注釋KEGG(Kyoto Encyclopedia of Genes and Genomes,京都基因與基因組百科全書)是基因組破譯方面的數據庫,在給出染色體中一套完整基因的情況下,它可以對蛋白質交互網絡在各種細胞活動起的作用作出預測,方便地尋找與行使某一類功能相關的所有注釋上的基因[16-17]。由圖5 可知,LY1 菌株的基因通過KEGG 分類主要分為5 大類,分別為細胞過程(Cellular Processes)、環境信息處理(Environmental Information Processing)、遺傳信息處理(Genetic Information Processing)、代謝(Metabolism)和有機系統(Organismal Systems),其中注釋到代謝的相關基因最多,達2 255 個,注釋到細胞過程、環境信息處理、遺傳信息處理和有機系統的基因數分別為99、613、334 和31 個。
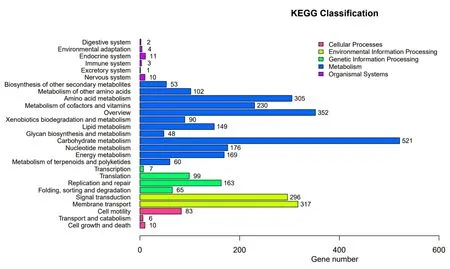
圖5 多粘類芽孢桿菌LY1 的KEGG 功能注釋
2.3.3 COG 注釋蛋白質直系同源簇數據庫(COG,Cluster of Orthologous Groups of Prot-eins)是用于同源蛋白注釋的數據庫,是將細菌、藻類和真核生物等完整基因組的編碼蛋白根據系統進化關系構建而成,蛋白編碼序列與之比對后可以預測蛋白的功能[18-19]。多粘類芽孢桿菌LY1 的COG 功能注釋結果如圖6 所示,注釋到碳水化合物轉運及代謝(Carbohydrate transport and metabolism)和轉錄(Transcription)分類的基因最多,分別為433(11.59%)和398 個(10.65%)。
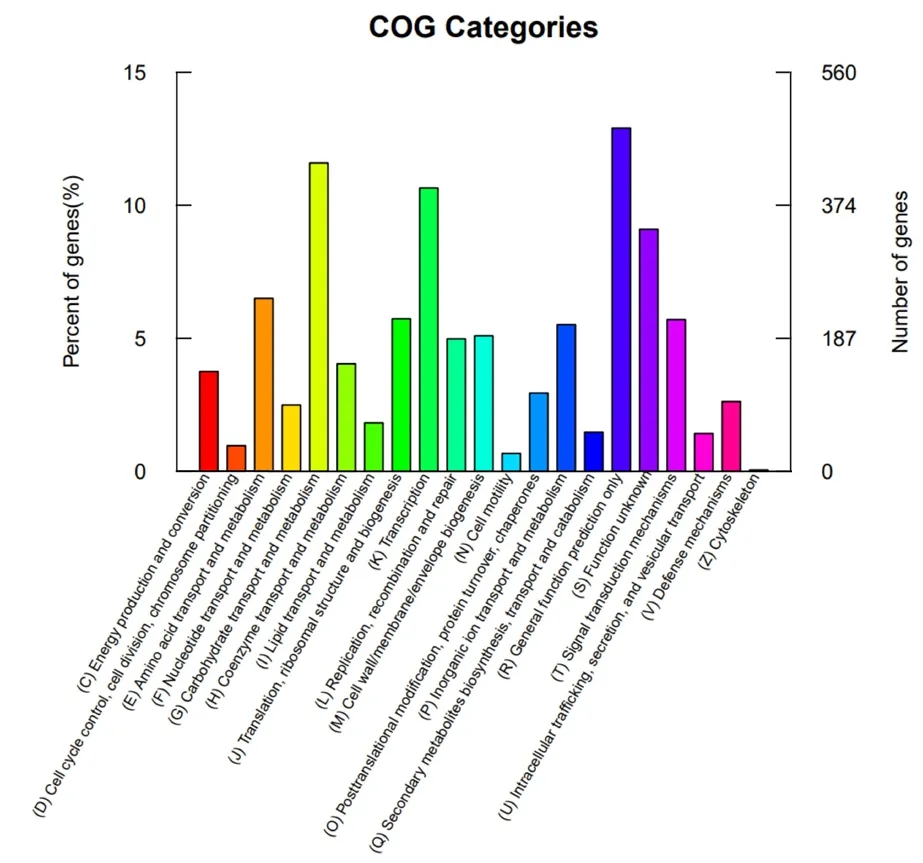
圖6 多粘類芽孢桿菌LY1 的COG 功能注釋
2.4 毒力因子分析
把菌株LY1 的基因蛋白序列在VFDB 致病因子數據庫(Virulence Factors of Pathogenic Bacteria)中進行比對,將基因與其對應的毒力因子(Virulence Factors,VF)功能注釋信息相結合,發現注釋到SetB 組(full dataset,共30 053 個與毒力因子相關的基因)的蛋白序列共326 個,占比5.61%;注釋到SetA 組(core dataset,共2 585 個與毒力因子相關的基因)的蛋白序列共301 個,占比5.18%(表1)。

表1 多粘類芽孢桿菌LY1 的基因組基本特征
2.5 系統發育進化分析
16S rRNA 基因是細菌上編碼rRNA 相對應的DNA 序列,存在于所有細菌的基因組中,具有高度的保守性和特異性,是病原菌檢測和鑒定的一種有效參照[20-21]。如圖7 所示,LY1 菌株與Paenibacillus polymyxaM1、Paenibacillus polymyxaOSY-DF、Paenibacillus polymyxaSC2、Paenibacillus peoriae、Paenibacillus jamilae、Paenibacillus polymyxaE681 等菌株的距離較近。
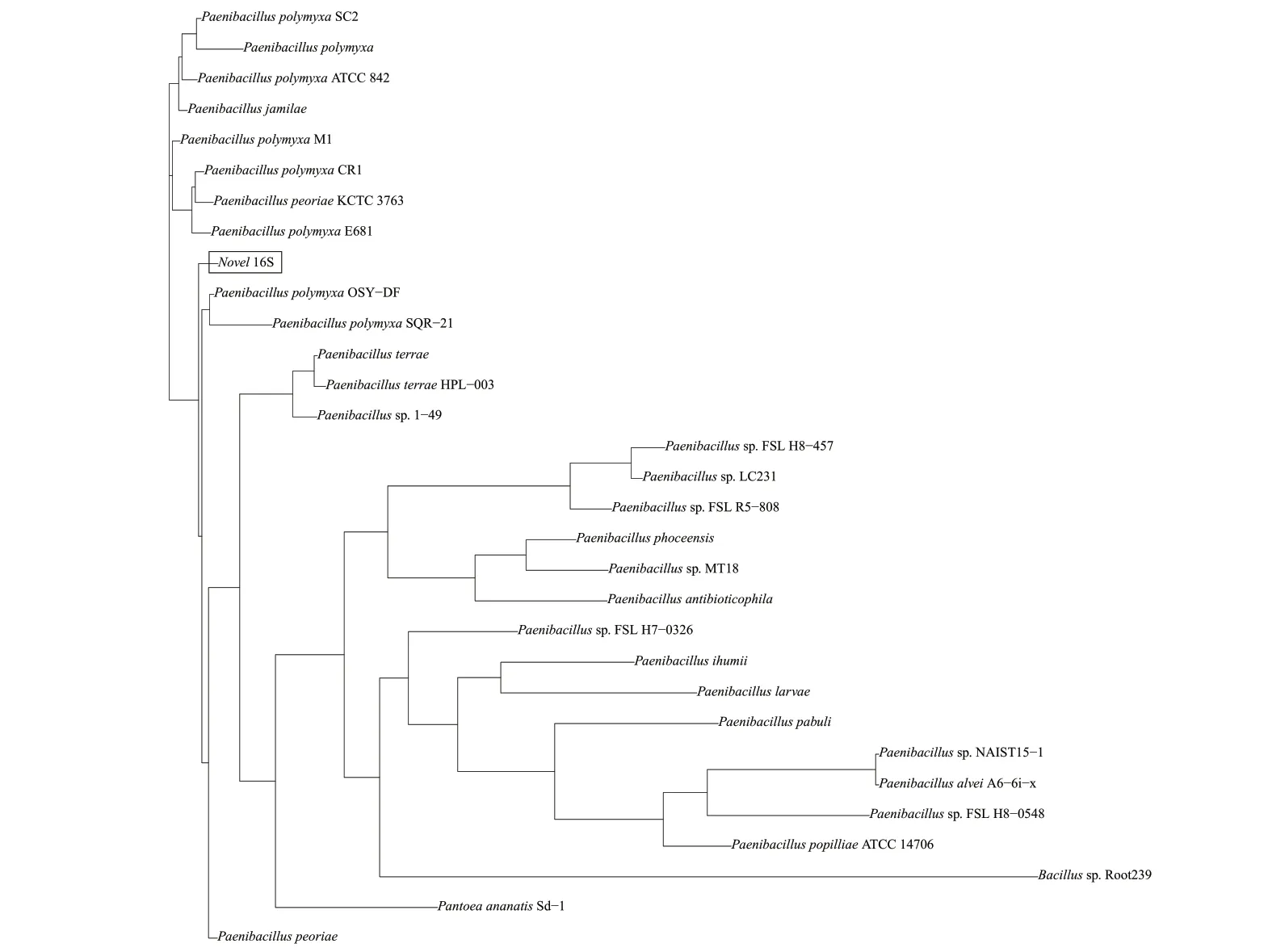
圖7 菌株LY1 基于16S rRNA 的系統發育樹
3 小 結
目前,人們對多粘類芽孢桿菌的功能有了較為全面地了解,但其分子作用機制以及遺傳工程改造等方面的研究報道還不多[3]。該研究通過全基因組測序分析了多粘類芽孢桿菌LY1 的基因組序列,獲得其基因組基本特征、基因功能注釋及分類、系統發育進化關系等關鍵信息,為其后續的功能挖掘、遺傳操作系統建立、基因工程改造等提供了研究基礎與前提條件。結果表明,多粘類芽孢桿菌LY1 的基因組長度為5 765 474 bp,共有6 086 個蛋白質編碼基因、162 個 tRNA 基因、42 個rRNA 基因,GC 含量約為45.23%;在GO 功能注釋結果中,在生物過程分類下,代謝過程和細胞過程中的基因占比最多,分別占比40.77%和38.98%;在細胞組分分類下,分類到細胞和細胞組成中的基因占比最多,為35.18%;在分子功能分類下,分類到催化活性和結合中的基因占比最多,分別為40.34%和35.78%;系統發育進化分析顯示,菌株LY1 與Paenibacillus polymyxaM1、Paenibacillus polymyxaOSY-DF、Paenibacillus polymyxaSC2、Paenibacillus polymyxaE681 等菌株進化距離較近。同時,根據多粘類芽孢桿菌LY1 的基因組信息及發育進化信息,筆者認為有望將其開發成新型生物防治制劑和農業肥料添加劑等在農業生產領域廣泛應用。
猜你喜歡
數學小靈通(1-2年級)(2021年4期)2021-06-09 06:25:56
大眾健康(2021年6期)2021-06-08 19:30:06
中學生數理化·七年級數學人教版(2019年4期)2019-05-20 10:06:32
中學生數理化·七年級數學人教版(2018年6期)2018-06-26 08:36:06
初中生世界·七年級(2017年9期)2017-10-13 22:27:46
財經(2017年15期)2017-07-03 22:40:49
財經(2017年2期)2017-03-10 14:35:35
財經(2016年15期)2016-06-03 07:38:02
財經(2016年3期)2016-03-07 07:44:46
財經(2016年6期)2016-02-24 07:41:51
