肌炎特異性抗體陽性的多發性肌炎/皮肌炎相關間質性肺疾病22例臨床特點分析
2022-07-04 10:46:42劉嘉林石伊寧王敏劉煒張妍蓓方浩徽
臨床肺科雜志 2022年7期
劉嘉林 石伊寧 王敏 劉煒 張妍蓓 方浩徽
結締組織病為ILD最常見病因。特發性炎性肌病(idiopathic inflammatory myopathies, IIM),是一種結締組織病,分為多發性肌炎(polymyositis, PM),皮肌炎(dermatomyositis, DM),免疫介導的壞死性肌病(immune-mediated necrotizing myopathy, IMNM)和包涵體肌炎(inclusion body myositis,IBM)[1]。PM和DM是IIM的最常見類型,約50%的IIM合并ILD[2-4]。部分PM/DM以ILD為首發表現或主要表現,亞急性甚至急性起病,極易誤診,死亡率較高。近年來[5],在PM/DM患者的血清中發現多種自身抗體,分為肌炎相關性抗體(myositis-associated autoantibody, MAA)和肌炎特異性抗體(myositis specific antibody, MSA),部分MSA可提示IIM的特征及預后,本研究回顧性分析了本院確診PM/DM-ILD的22例患者的臨床特點,探討如何結合臨床表現與肌炎抗體檢測等手段,對PM/DM-ILD進行早期診斷、分類及預后評估,并探討肌炎抗體檢測在ILD診療中的意義。
資料與方法
一、研究對象
納入安徽省胸科醫院2019年4月-2021年6月收治的22例明確診斷為多發性肌炎/皮肌炎相關間質性肺疾病患者,間質性肺疾病參照 2013 年美國胸科學會/歐洲呼吸病學會(ATS/ERS)的診斷標準[6],所有患者均符合1975年Bohan/Peter標準[7]/2010中國PM和DM診治療指南[8]/EULAR/ACR2017年IIM分類標準[1],分為抗合成酶綜合征相關間質性肺疾病組(ASS-ILD)13例、抗MDA5陽性相關間質性肺疾病(MDA5-ILD)5例、其他PM/DM相關間質性肺疾病組(4例)。三組一般情況比較,差異無統計學意義(P>0.05),具有可比性。該研究通過安徽省胸科醫院倫理委員會審核(K2021-010)并批準。
二、納排標準
納入標準:(1)年齡在18歲~80歲之間;(2)經臨床表現、胸部HRCT、肺功能和(或)病理活檢確診ILD;(3)肌炎特異性抗體檢測結果至少一項中等及中等以上陽性,并結合臨床表現和其他實驗室檢查可明確診斷PM/DM;(4)病史及臨床資料基本完整。
排除標準:(1)合并其他結締組織病如類風濕關節炎(RA)、系統性紅斑狼瘡(SLE)、系統性硬化癥(SSc)、干燥綜合征(pSS)、ANCA相關性血管炎(AAV)等其他可引起ILD的患者;(2)其他已知病因的ILD患者,如塵肺、藥物引起的間質性肺炎、放射性肺炎等;(3)合并肺部或其他系統惡性腫瘤;(4)合并心腎功能不全;(5)病史及臨床資料不全。
三、臨床資料和方法
1 一般資料
對病歷資料進行回顧性分析,包括:患者的人口學資料,臨床癥狀體征(發熱、肌痛、肌無力、技工手、雷諾現象、gottron征、眶周紫斑),各項實驗室檢查(肌酶譜、紅細胞沉降率、C反應蛋白、抗核抗體譜、ANCA抗體四項、肌炎抗體譜等)。
2 胸部HRCT表現
根據影像學表現分為:OP型、NSIP型、NSIP+OP型、UIP型等。
3 肺功能
檢測指標包括:肺活量(VC)、用力肺活量(FVC)、第1秒用力呼氣容積(FEV l)、第1秒用力呼氣容積/用力肺活量(FEV l/FVC)和一氧化碳彌散量(DLCO)。
4 療效判斷
出院前評估療效,統計病情好轉、穩定、惡化和死亡情況。好轉的標準:影像學病灶吸收≥25%和/或肺功能通氣、彌散功能改善。穩定的標準:影像學病灶吸收<25%和/或肺功能通氣、彌散功能無明顯惡化。惡化的標準:影像學病灶范圍增大和/或肺功能通氣、彌散功能下降。
5 隨訪時間治療后3月。
四、統計學分析
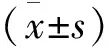
結 果
一、一般情況
本研究納入的22例明確診斷為多發性肌炎/皮肌炎相關間質性肺炎患者,男10例(45.5%),女12例(54.5%),其中ASS-ILD組男5例,女8例,該組平均年齡56.7±11.6歲,MDA5-ILD組男1例,女4例,該組平均年齡57±9.75歲,其他PM/DM相關間質性肺疾病組男4例,女0例,該組平均年齡66±8.04歲
二、臨床特征
1 癥狀與體征
技工手、雷諾現象、皮膚癥狀(Gottron征、眶周紫斑)、肌無力癥狀等的陽性率在三組間差異有統計學意義(P<0.05)。進一步比較顯示,ASS-ILD組更容易出現技工手及雷諾現象,而發熱、肌無力癥狀、首發皮膚表現(Gottron征、眶周紫斑)發生率MDA5組明顯升高。肌酸激酶(CK)方面,其他PM/DM相關間質性肺疾病組升高更明顯(表1)。

表1 3組患者臨床特征比較
2 胸部HRCT特征
22例患者中,11例(50.0%)表現為機化性肺炎(organizing pneumonia, OP),8例(36.4%)表現為非特異性間質性肺炎(non-specific interstitialpneumonia,NSIP),1例(4.5%)表現為NSIP合并OP,2例(9.1%)表現為普通型間質性肺炎(usual interstitial pneumonia,UIP)(表2,圖1)。

表2 3組患者影像學特征比較[n/%]
3 肺功能特征
肺功能檢查結果顯示,在22例患者中,均表現為限制性通氣功能障礙及彌散功能障礙。
4 呼吸衰竭情況
MDA5-ILD組發生率最高(80%),其他組次之(50%),ASS-ILD組最低(23.1%)。
5 治療與轉歸情況
其中在22例患者中,17例(77.3%)患者使用激素聯合免疫抑制劑(環磷酰胺、他克莫司)治療,3例(13.6%)患者僅予激素治療,2例(9.1%)患者未治療。22例患者中,共6例患者死亡,其中ASS-ILD組死亡1例(7.7%),MDA5組死亡4例(80.0%),其他組死亡1例(25%),存活的16例(72.7%)。患者中10例轉歸為好轉,5例穩定,1例惡化,三組患者轉歸差異與既往文獻報道一致,但差異無統計學意義(P>0.05)(表3,表4)。

表3 3組患者治療方式比較[n/%]

圖1A ASS-ILD組患者NSIP影像學表現;圖1B ASS-ILD組患者NSIP+OP影像學表現;圖1C MDA5-ILD組患者OP影像學表現;圖1D 其他組患者UIP影像學表現。

表4 3組患者預后比較[例/%]
討 論
間質性肺疾病可分為1)已知原因ILD,2)特發性間質性肺炎,3)肉芽腫病,4)其他罕見ILD。既往文獻報道結締組織病約占ILD的30%~50%,IIM是一組以四肢近端肌肉受累為主要表現的結締組織病,PM/DM占IIM的70%左右,PM/DM易引起ILD,且部分為快速進展的ILD,近年來發現的肌炎特異性抗體因其對PM/DM高度的特異性,可用于PM/DM的早期診斷、分類及評估其預后。
通過回顧性分析發現,不同肌炎抗體陽性的PM/DM-ILD臨床特征存在明顯差異。在22例肌炎特異性抗體陽性的ILD患者中,ARS抗體(59.1%)及MDA5抗體(22.7%)發生率最高,既往多項研究表明[9-10],ARS抗體為PM/DM中最常出現的抗體,其次為抗MDA5抗體,但受人種、地域、環境等影響,其余抗體的分布國內外存在一定差異。在我們的研究中的ASS-ILD組患者易出現技工手及雷諾現象,MDA5-ILD組患者突出表現則為肌無力癥狀和首發皮膚表現(Gottron征、眶周紫斑),有文獻表明[11],抗 Jo-1 抗體合并ILD發病率高達70%~90%。吳燕芳等人[12]報道了在15例抗Jo-1抗體綜合征患者中ILD發病率高達100%。此外,還有研究發現ILD、技工手、雷諾現象等臨床表現與抗合成酶綜合征密切相關[13],而MDA5陽性患者易出現Gottron征,眶周紫斑[14],與本研究結果基本相符。
CTD-ILD最常見的影像學及病理表現均為NSIP[15-16],而PM/DM-ILD最常見的影像學表現文獻報道不一,但最常見兩種類型的均為NSIP及OP[17-18],本研究的22例患者中,最常見的影像學表現分別為機化性肺炎(OP),占50.0%,其次為非特異性間質性肺炎(NSIP),占36.4%,1例(4.5%)表現為NSIP合并OP,2例(9.1%)表現為普通型間質性肺炎(UIP),值得注意的是,MDA5-ILD組的5例患者全部為OP型,且其中4例為快速進展的間質性肺疾病(RP-ILD),這與既往報道亞裔MDA5陽性患者易出現以OP為主要影像/病理學表現的RP-ILD符合[19]。
PM/DM因侵犯骨骼肌,故可引起肌酶升高,不同肌炎特異性抗體陽性的ILD患者,肌酸激酶(CK)升高亦有所不同,值得注意的是,在MDA5-ILD組無1例CK升高,考慮原因為抗MDA5陽性患者為無肌病型皮肌炎(CAMD)最常見類型,一般無肌炎表現,故很少出現肌酶升高[20-21]。
經過住院期間觀察及后續隨訪,22例患者死亡6例,死因主要為呼吸衰竭、合并感染、放棄治療等,具體分析發現ASS-ILD的患者預后良好,而MDA-5-ILD的患者預后差。來自Hozumi等[16]的研究表明,ARS陽性的PM/DM-ILD患者10年生存率明顯好于ARS陰性的PM/DM-ILD患者(91.6%比58.7%),Rohit Aggarwal等人[22]對202名抗合成酶抗體的患者進行了長達24 年的回顧性評估,發現Jo-1陽性患者的 5年生存率為 90%,Jo-1陰性患者為 79%。另外研究發現亞洲人群抗MDA5陽性患者易出現快速進展的ILD,MDA5陽性為ILD獨立危險因子,MDA5陽性ILD患者總體預后差,生存時間短[23-25],因此,可以看出肌炎特異性抗體對于患者預后評估是極為重要的。
雖然大多數PM/DM具有特征性皮膚表現,但仍有相當一部分PM/DM患者以ILD為首發表現或主要表現,亞急性甚至急性起病,此類病人多首診呼吸科,由于PM/DM-ILD常見的影像學表現(NSIP及OP)與各種感染性疾病如病毒性肺炎、大葉性肺炎的影像學表現有一定相似之處,尤其是NSIP的特點與新型冠狀病毒肺炎(COVID-19)早期表現非常類似,加上很多呼吸科醫生對結締組織病認識不足,導致該類患者常常未能及時進行肌炎特異性抗體等檢查,卻被誤診為各種感染性疾病,從而貽誤了最佳治療時機,甚至導致患者病情惡化、死亡[26-27]。本研究的22例患者中近一半患者初始診斷為“肺部感染”“肺部陰影”“機化性肺炎”等疾病,后經進一步檢查方診斷為PM/DM-ILD,充分說明肌炎抗體的檢測在ILD患者特別是以NSIP或OP為主要影像學表現的患者的必要性。
綜上所述,本研究證實了肌炎抗體的檢測特別是肌炎特異性抗體的檢測對ILD的病因診斷、分類、預后評估具有重要意義,對病因不明的ILD患者,特別是影像學表現為OP、NSIP的患者,應盡早進行肌炎抗體篩查,從而避免漏診、誤診,并讓患者能盡早被診治,盡早獲益。本研究的局限性無疑是樣本量相對較小,擬在后續進行樣本量更大且更為深入的研究進一步分析各種類型PM/DM-ILD的臨床差異及轉歸情況。
