基于合成路線分析的鹽酸阿托莫西汀有關(guān)物質(zhì)研究進展
2022-07-09 03:15:04李樹英倪志偉張慶盧大峰臧淑超王玉玲翟光喜
藥學研究 2022年6期
李樹英,倪志偉,張慶,盧大峰,臧淑超,王玉玲,翟光喜
(1.山東大學藥學院,山東 濟南 250012;2.青島雙鯨藥業(yè)股份有限公司,山東 青島 266000)
鹽酸阿托莫西汀,化學名為(R)-N-甲基-3-(2-甲基苯氧基)-3-苯丙基胺鹽酸鹽,結(jié)構(gòu)式如圖1所示,為一種選擇性去甲腎上腺素攝取抑制劑,是由Eli Lilly公司研制開發(fā)的新一代治療兒童注意缺陷多動障礙(attention deficit hyperactivity disorder,ADHD)的非中樞神經(jīng)興奮劑[2-3],可以用于治療所有年齡段的ADHD患者[1]。本品已于2002年7月獲美國食品藥品監(jiān)督管理局(FDA)批準,于2003年1月在美國上市,已上市的劑型包括膠囊劑以及口服溶液劑兩種,是FDA許可用于治療ADHD的第一個非中樞神經(jīng)興奮劑,為提高其用藥順應性,有文獻先后對鹽酸托莫西汀口服溶液[4-5]、鹽酸托莫西汀片速溶片[6]、口崩片[7]、口腔速溶膜劑[8]進行了系統(tǒng)研究。
雜質(zhì)控制是藥品安全性得到有效控制的關(guān)鍵手段,而基于合成路線分析是雜質(zhì)控制的重要策略,可以通過對藥品中的每一個雜質(zhì),從源頭追溯,可以較為全面地依據(jù)其生理活性制定相應的限度要求,以便加深對藥品各雜質(zhì)產(chǎn)生原因的理解,為工藝優(yōu)化、處方開發(fā)、包裝形式、存儲運輸?shù)忍峁┲匾募夹g(shù)支持[9-10]。藥品的雜質(zhì)可能來源于起始物料、試劑、配位體、催化劑、中間體以及副產(chǎn)物等合成、精制工藝過程,也可能來源于儲存、運輸以及制劑生產(chǎn)中產(chǎn)生的降解雜質(zhì)[11-13],對于藥品的雜質(zhì)進行全面剖析,需要一條主線貫穿整個過程,以防漏掉或分析不全面現(xiàn)象的出現(xiàn),雜質(zhì)譜便是這條主線。近年來,特別是因沙坦類藥物中的亞硝胺類遺傳毒性雜質(zhì)引發(fā)了國內(nèi)對于基因毒性雜質(zhì)研究的關(guān)注[14-16],雜質(zhì)譜研究日益引起了藥學工作者以及藥品監(jiān)管者的重視。
隨著鹽酸阿托莫西汀膠囊及口服溶液在臨床應用地逐步推廣以及其他新劑型的開發(fā),特別是對于兒童用藥,臨床用藥安全性日益重要,本文對已有的鹽酸阿托莫西汀及其制劑有關(guān)物質(zhì)文獻報道進行全面歸納分析,從合成路線的角度,全面系統(tǒng)地對鹽酸阿托莫西汀的有關(guān)物質(zhì)進行了總結(jié),以期為該產(chǎn)品的質(zhì)量控制,特別是有關(guān)物質(zhì)的控制提供參考。
1 鹽酸阿托莫西汀有關(guān)物質(zhì)分析
對鹽酸阿托莫西汀的合成路線進行檢索可知[17],已產(chǎn)業(yè)化的合成路線均是以3-甲胺基苯丙醇為起始物料,在堿性條件下與鄰氟甲苯經(jīng)醚化、拆分、成鹽得到[18-19],結(jié)合EP9.0、USP41藥典中鹽酸阿托莫西汀質(zhì)量標準以及文獻報道[20-21],從起始物料、拆分劑、中間體以及副產(chǎn)物等合成、精制工藝過程以及可能存在的降解路徑進行了分析,鹽酸阿托莫西汀可能的有關(guān)物質(zhì)如圖1所示。

圖1 鹽酸阿托莫西汀的合成路線及相關(guān)雜質(zhì)
由圖1可知,在合成過程中由于拆分過程中存在可能產(chǎn)生S-異構(gòu)體B以及拆分試劑C的殘留、起始物料A的殘留等雜質(zhì);同時對于醚化試劑鄰氟甲苯,由于其自身存在的位置異構(gòu)體(間氟甲苯、對氟甲苯)以及工藝雜質(zhì)(氟苯、2,6-二氟甲苯),導致在鹽酸阿托莫西汀合成過程中因起始物料引入相應的工藝雜質(zhì)D、E、F、G、H;且據(jù)報道[17-18]鹽酸阿托莫西汀在硫酸作用下,可降解為雜質(zhì)A、I;在高氯酸強酸性、強氧化作用下可降解為J、K。基于圖1的鹽酸阿托莫西汀合成路線及相關(guān)雜質(zhì)分析,鹽酸阿托莫西汀及其制劑中可能存在的各雜質(zhì)結(jié)構(gòu)及溯源如表1所示。
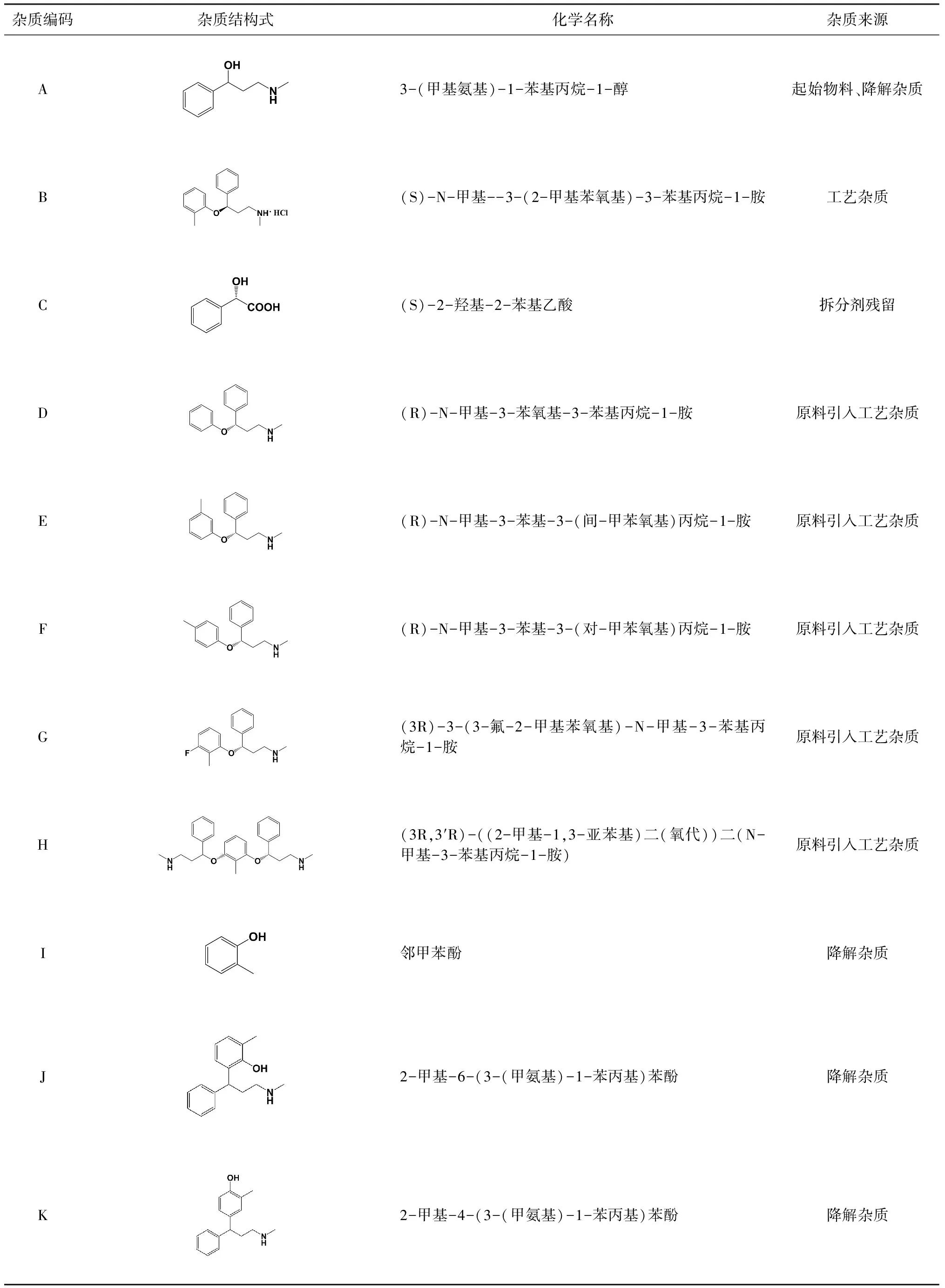
表1 鹽酸阿托莫西汀各雜質(zhì)結(jié)構(gòu)、名稱及來源信息
2 正相高效液相色譜法
由圖1可知:雜質(zhì)E、F與主成分互為同分異構(gòu)體,由起始物料鄰氟甲苯中的雜質(zhì)間氟甲苯、對氟甲苯引入的工藝雜質(zhì);雜質(zhì)B與主成分為對映異構(gòu)體,是拆分的副產(chǎn)物殘留引入。上述3個雜質(zhì)均與主成分分子式一致,且結(jié)構(gòu)相似,常規(guī)的反相高效液相法較難分離完全。
對于雜質(zhì)B、E、F 3個雜質(zhì)的分離《歐洲藥典》EP9.0以及《美國藥典》USP41-NF36均采用纖維素衍生物硅膠柱,以二乙胺-三氟乙酸-異丙醇-正庚烷(1.5∶2.0∶150∶846.5)為流動相,雜質(zhì)B與D的系統(tǒng)適應性實驗中分離度大于1.8,亦能滿足對映異構(gòu)體B、同分異構(gòu)體E、F的分離要求。
Eli Lilly公司的Sellers等[22]系統(tǒng)研究了雜質(zhì)B、D、E、F之間以及各雜質(zhì)與主成分之間的色譜分離方式,采用非水系多糖手性柱、磺化β-環(huán)糊精改性辛基柱以及HS-β-CD(高磺化β-環(huán)糊精,七-6-磺化β-環(huán)糊精)為手性選擇劑的毛細管電泳法三種方法進行分離實驗,結(jié)果表明,采用Chiralcel OD-H柱,以正庚烷/異丙醇/二乙胺/三氟乙酸 (85/15/0.15/0.2,V/V/V/V)為流動相,各雜質(zhì)間以及各雜質(zhì)與主成分之間的分離度能夠滿足要求,優(yōu)化后最終確定的色譜圖如圖2所示。

圖2 各雜質(zhì)在色譜條件(色譜柱:Chiralcel OD-H,4.6 mm×250 mm,5 μm;流動相:正庚烷/異丙醇/二乙胺/三氟乙酸 (85/15/0.15/0.2,V/V/V/V);1.0 mL·min-1;檢測波長:273 nm;柱溫:室溫)的分離情況,其中雜質(zhì)V為消旋體
陳杰等[23]采用新型的硅膠鍵合聚甲基丙烯酸酯手性柱CHIRLPAK?OT (+),流動相為甲醇-異丙醇-三乙胺(960∶40∶0.5),檢測器為蒸發(fā)光散射檢測器,漂移管溫度為90 ℃,氣流速度為2.5 L·min-1,該色譜柱具有傳統(tǒng)硅膠正相柱的分離特點,但又不具有后者對水高度敏感的特性,且與后者比較另具有手性分離能力。同時將紫外二極管陣列檢測器(DAD)串聯(lián)蒸發(fā)光散射檢測器(ELSD),利用ELSD能夠檢出無紫外吸收的雜質(zhì)的特點,比較兩種檢測器檢出雜質(zhì)譜的差異,從而建立了鹽酸阿托莫西汀中包括旋光異構(gòu)體及同分異構(gòu)體在內(nèi)的有關(guān)物質(zhì)的檢查方法。
吳擁軍等[24]采用Chiralcel OD(4.6 mm×250 mm,5 μm)手性柱,以正己烷-異丙醇-二乙胺(80∶20∶0.2)為流動相對對映異構(gòu)體的分離進行了研究,結(jié)果表明左旋鹽酸阿托莫西汀峰和右旋鹽酸阿托莫西汀峰能很好地分開,分離度為5.5,拖尾因子小于1.2,理論塔板數(shù)大于5 000,可以用于原料藥中S-鹽酸阿托莫西汀含量的測定。
孫冬雪等[25]采用硅膠表面涂敷有直鏈淀粉-三(3,5-二甲苯基氨基甲酸酯)為填料的手性色譜柱,以一定比例的正己烷低級醇堿性添加劑為流動相,建立了一種可以有效分離雜質(zhì)B、D、E、F的分離方法。
3 反相高效液相色譜法
由圖1可知,除B、D、E、F 4種雜質(zhì)外,其他各雜質(zhì)在結(jié)構(gòu)上存在一定的差異,可以采用一般的反相高效液相色譜進行分離控制。
Gavin等[20]以質(zhì)量源于設(shè)計(Quality by Design,QbD)的理念,對鹽酸阿托莫西汀中的雜質(zhì)A、B、C、D、F、G、H、I(其中D、F、G、H為消旋體)以及其它6個雜質(zhì)進行了系統(tǒng)研究,采用離子對-液相色譜法,以拖尾因子、運行時間、分離度[RS(A-C)、RS(D-主成分)、RS(G-主成分)]以及柱壓為評價指標,考察緩沖鹽濃度、pH值、離子度濃度、正丙醇比例以及柱溫的影響,如圖3所示,在此基礎(chǔ)上得到了最佳的色譜條件,并進行了全面的分析方法學驗證。

圖3 緩沖鹽濃度、pH值、離子度濃度、正丙醇比例以及柱溫對拖尾因子、運行時間、分離度[RS(A-C)、RS(D-主成分)、RS(G-主成分)]以及柱壓的影響。其中:Rs(1-2)為RS(A-C)、Rs(3-4)為RS(D-主成分)、Rs(5-4)為RS(G-主成分)
呂和平等[26]采用Elclipse XDB-C8(4.6 mm×150 mm,3.5 μm)色譜柱,以正丙醇:緩沖液(2.9 g·L-1磷酸用1 000 mL水溶解,用5 mol·L-1KOH調(diào)pH值至2.5,再加入5.9 g辛烷磺酸鈉)- (27∶73) 為流動相,檢測波長為215 nm,雜質(zhì)A、E、H三者之間以及三者與主成分之間均能夠有效分離,且經(jīng)強制破壞無干擾,建立一種可以有效控制鹽酸阿托莫西汀原料藥有關(guān)物質(zhì)的分析方法。
張小燕等[27]采用Kromasil C18(4.6 mm×250 mm,5 μm) 色譜柱,流動相:乙腈-水-三乙胺溶液(40∶60∶0.5,冰醋酸調(diào)pH至6.0),流速為1.0 mL·min-1,檢測波長為270 nm,柱溫為室溫,對鹽酸阿托莫西汀的有關(guān)物質(zhì)進行了測定。雖經(jīng)強制降解試驗進行專屬性考察,但未對特定雜質(zhì)進行定位研究。
何淑旺等[28]在建立的高效液相色譜(HPLC)法測定鹽酸阿托莫西汀原料藥的含量方法中,對雜質(zhì)A、E、F 3個工藝雜質(zhì)進行了定位,可以采用該方法對雜質(zhì)A、E、F進行分離,由于該方法用于含量的測定,未對其他雜質(zhì)的分離情況進行相關(guān)研究。
Gavin等[19]對鹽酸阿托莫西汀的起始物料3-(甲基氨基)-1-苯基丙烷-1-醇進行了系統(tǒng)研究,針對不同合成路線所引入的雜質(zhì),采用親水相互作用色譜法(HILIC)、離子對-液相色譜法、離子相互作用色譜法對6條路線引入的12個雜質(zhì)(如圖4所示)進行了分離研究,并通過氣質(zhì)色譜以及液質(zhì)色譜進行了結(jié)構(gòu)確認。

圖4 鹽酸阿托莫西汀起始物料3-(甲基氨基)-1-苯基丙烷-1-醇合成路線及相關(guān)雜質(zhì)
張小燕等[29]以2,3,4,6-四-O-乙酰基-β-D-葡萄糖異硫氰酸酯(GITC)為柱前手性衍生化試劑,采用Agilent Zorbax SB-C18(4.6 mm×250 mm,5 μm)色譜柱,以乙腈-四氫呋喃-水(體積比為41∶14∶45)為流動相,流速1.0 mL·min-1;檢測波長254 nm,柱溫35 ℃。在優(yōu)化試驗條件下,鹽酸阿托莫西汀與S-異構(gòu)體的分離度R>1.5,避免手性柱成本高、壽命短、通用性差等缺點,建立了一種柱前衍生化反相高效液相色譜(RP-HPLC)法測定鹽酸阿托莫西汀中S-異構(gòu)體雜質(zhì)的方法。
4 小結(jié)
有關(guān)物質(zhì)作為貫穿藥品雜質(zhì)研究的主線,可以從起始物料、中間體、催化劑、副產(chǎn)物等工藝雜質(zhì)以及可能存在的降解途徑入手,對藥品有機雜質(zhì)譜進行全面剖析。通過對雜質(zhì)結(jié)構(gòu)的分析,對不同結(jié)構(gòu)的雜質(zhì)進行分門別類,可以為后續(xù)的分析方法建立提供參考。鹽酸阿托莫西汀作為FDA批準治療ADHD的第一個非中樞神經(jīng)興奮劑藥物,在臨床上得到了廣泛應用,本文通過對其有關(guān)物質(zhì)的全面剖析,并依據(jù)合成路線涉及的有關(guān)物質(zhì)對國內(nèi)外的質(zhì)控研究進行了歸納總結(jié),針對同分異構(gòu)體難以分離難題,可以采取不同的控制策略,以正相色譜以及反相色譜相結(jié)合的方式可以較為全面地加以有效控制,但是在分析過程中存在交叉檢測情景,需要針對性加以分析,以最大的檢測能力以及檢測結(jié)果作為選擇依據(jù)。通過本文對于鹽酸阿托莫西汀有關(guān)物質(zhì)及質(zhì)控情況的歸納總結(jié),為該品種系列制劑的有關(guān)物質(zhì)的質(zhì)控提供參考,確保兒童臨床用藥安全性。
