顯性營養不良型大皰性表皮松解癥COL7A1基因突變分析
2022-07-13 13:59:38謝錦瑩周順婷黃閩嘉羅志強周書文
中國麻風皮膚病雜志 2022年9期
祝 玉 吳 瑋 謝錦瑩 周順婷 黃閩嘉 羅志強 周書文
廣東醫科大學附屬醫院皮膚科,廣東湛江,524000
營養不良型大皰性表皮松解癥(epidermolysis bullosa,EB)是一組以皮膚和(或)黏膜脆性增加為特征的罕見遺傳病,可發生于局部或全身。它是由編碼不同蛋白質的基因突變引起的,這些蛋白質主要與真皮-表皮連接的結構和功能有關。根據皮膚裂隙水平(從上到下)分為四種類型:單純型(EBS,epidermolysis bullosa simplex)、交界型(JEB,junctional epidermolysis bullosa)、營養不良型(DEB)和Kinder綜合征(KS)。分子遺傳學表明,位于染色體3p21的COL7A1基因(C7)為DEB的致病基因[1]。本文通過基因測序確診2例來自一個廣東DEB家系的患者,并發現了位于86號外顯子的基因突變位點。
1 臨床資料及家系調查
先證者,女,廣東廉江人,51歲。雙脛前反復結節、水皰,伴瘙癢40余年就診。患者自10歲以來雙脛前反復出現丘疹和水皰,慢慢演變成結節和色素沉著,瘙癢明顯,逐漸雙上肢也出現類似皮損。皮膚科情況:雙側小腿見密集分布孤立暗紅色結節,部分中央結痂,見少部分丘疹,無水皰。雙側大腿可見散在紅色丘疹、結節,部分中央結痂,無水皰。雙上肢可見散在紅色丘疹,部分表面結痂。趾甲可見甲增厚、表面凹凸不平和甲萎縮(圖1)。組織病理:正角化過度,表皮略肥厚,表皮突消失,見表皮下裂隙,真皮淺層膠原纖維增生致密,見少許淋巴組織細胞和漿細胞浸潤(圖2)。免疫熒光:基底膜帶IgG、IgA、IgM及C3均陰性。初步診斷為大皰性表皮松解癥。予阿維A膠囊20 mg日2次,復方甘草酸苷片50 mg日3次,復方醋酸曲安奈德溶液及鹵米松/三氯生乳膏外用治療,效果不佳,目前正在隨訪中。
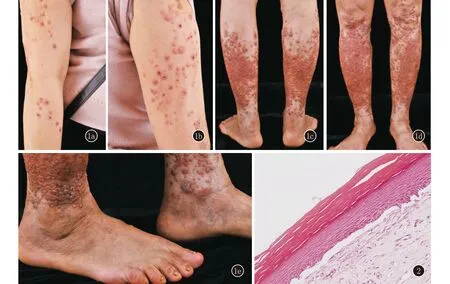
圖1 先證者臨床照片 1a、1b:雙上肢散在多發丘疹,部分中央結痂,伴脫屑;1c、1d:雙側小腿可見密集分布紅色丘疹、結節,部分融合。雙側大腿可見散在多發紅色丘疹,部分中央結痂;1e:趾甲可見甲增厚、表面凹凸不平和甲萎縮 圖2 先證者組織病理:正角化過度,表皮突消失,見表皮下裂隙,真皮淺層膠原纖維增生致密(HE,×200)
先證者胞妹,44歲,雙下肢丘疹、結節30余年,伴輕微瘙癢。患者自10歲以來雙下肢丘疹、結節,伴輕微瘙癢。皮膚科情況:雙側小腿和足背可見密集分布類圓形丘疹、結節,部分呈線狀排列,背部和右側肘關節伸側見散在多發皮色扁平丘疹,無水皰。10趾甲可見甲增厚、表面凹凸不平和甲萎縮(圖3)。
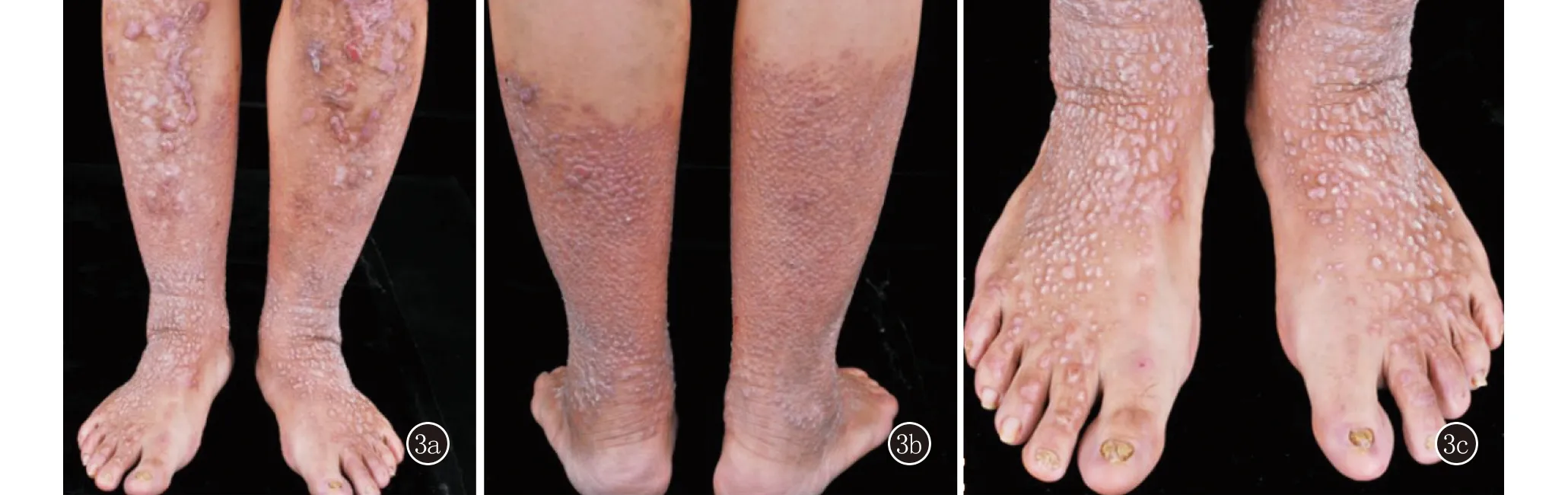
3a、3b:雙側小腿密集分布類圓形丘疹、結節,部分融合成線狀,雙側脛前可見片狀色素減退;3c:雙足背可見密集分布類圓形丘疹、結節,雙側趾甲可見甲營養不良
先證者的父親和胞妹的兒子亦有類似癥狀,母親、其余兩個姐妹和其子女無類似癥狀。該家系4代20名成員,發現有4例患者(圖4),父母否認近親結婚。根據家系調查結果,該病的遺傳方式為常染色體顯性遺傳。

4a:家系圖(4代20名成員,發現有4例患者,分別為先證者及其父親、妹妹和外甥),箭頭示先證者;4b:先證者及其胞妹測序分析提示:雜合突變,位于外顯子86上的c.6761G>A;p.Gly2254Glu(上方為先證者,下方為其胞妹)
2 基因檢測
2.1 經患者知情同意后,取患者及其胞妹的外周靜脈血各6 mL,標本送至深圳華大臨床檢驗中心,采用芯片捕獲高通量方法對先證者進行DNA基因全外顯子直接測序。檢測區域:人類基因組中約2萬個基因的外顯子區;檢測策略:針對受檢者主訴,對OMIM(0nline Mendelian Inheritance in Man)數據庫收錄的明確致病關系基因進行分析;檢測方法:芯片捕獲高通量測序。
2.2 對先證者及其胞妹的外周血DNA用Sanger測序驗證突變位點。
3 結果
3.1 先證者 在先證者COL7A1基因外顯子86上檢出一個與受檢者臨床表型相關的意義未明變異c.6761G>A;p.Gly2254Glu(圖4),根據ACMG(American College of Medical Genetics and Genomics)指南,該變異被判斷為意義未明變異。采用Sanger測序驗證突變位點的結果顯示為一雜合突變。
3.2 患病胞妹 采用Sanger測序驗證突變位點的結果與先證者一致:c.6761G>A;p.Gly2254Glu,為雜合突變。
4 討論
EB是指皮膚或黏膜受到輕微外傷后即可引起水皰的一組異質的罕見遺傳性皮膚病,包括三個主要類型(單純型EB、交界型EB、營養不良型EB)和一種罕見變異型(Kinder綜合征,即肢端角化性皮膚異色病),共分為至少20種臨床亞型。角蛋白5和7、層粘連蛋-332、Ⅶ型膠原、大皰性類天皰瘡抗原2、網格蛋白、整合素等7個靶蛋白中任何一個基因突變均可導致一種EB的發生。目前為止,已經發現的可導致EB發生的基因有19個,其中編碼Ⅶ型膠原的COL7A1基因突變導致營養不良型EB的發生[2]。營養不良型EB分為常染色體顯性遺傳和常染色體隱性遺傳,顯性營養不良型EB(dominant dystrophic epidermolysis bullosa,DDEB)是由于Ⅶ型膠原蛋白發生負顯性突變,突變點位于蛋白中糖基結合點,并不導致Ⅶ型膠原蛋白的截斷,只是導致皮膚結構的異常,比如皮膚脆性增加、錨原纖維數量異常等。相反,隱性營養不良型EB(recessive dystrophic epidermolysis bullosa,RDEB)是Ⅶ型膠原蛋白的復合雜合子突變,提前出現的終止密碼子導致蛋白的截斷,從而導致Ⅶ型膠原的合成不穩定。從分子水平上看,本患者為編碼Ⅶ型膠原的COL7A1基因外顯子86上的錯義點突變,沒有導致Ⅶ型膠原蛋白的截斷,家系遺傳圖譜也顯示為常染色體顯性遺傳,符合DDEB的診斷。
與RDEB相比,DDEB一般臨床表現較輕微[3],多在嬰兒早期或兒童期發病,水皰及大皰多位于四肢伸側,尤其是關節部位,尼氏征常為陰性,愈后形成粟丘疹和瘢痕。因為是蛋白合成異常所致,不是自身抗體的原因,因此免疫熒光是陰性的。DDEB分為以下亞型:肢端型、脛前型、癢疹型、僅指甲受累型及新生兒大皰性表皮松解型,其變異型包括白色丘疹樣型和增生型。其中癢疹型DDEB表現為劇烈瘙癢性的紫紅色丘疹和結節,輕度水皰或糜爛,呈線狀排列,主要發生在脛前、前臂,少數發生在軀干,以及趾甲的萎縮變性,嬰兒或兒童期發病。與本例兩姐妹均在10歲左右發病,皮疹分布部位和皮疹特點都是相符的。結合臨床、病理(表皮下裂隙,免疫熒光陰性)和分子遺傳特點,符合癢疹型DDEB。
Masunaga等[4]總結了DEB的突變位點,提出隱性病例的突變分布于從N端到C端的整個VII型膠原。而與隱性病例相比,顯性病例的突變位于COL7A1基因的末端,主要為甘氨酸替代突變。本例患者基因檢測顯示為甘氨酸被谷氨酸替代,與文獻報道相符。本文中先證者及其胞妹COL7A1基因存在第6761位堿基G→A突變,導致第2254位甘氨酸替代,從而影響Ⅶ型膠原三螺旋結構區Gly-X-Y重復序列,破壞了三螺旋結構的穩定性。Clin Var數據庫記錄有該位點信息(c.6761G>A;p.Gly2254Glu),并評估為致病性變異。目前為止,只有2例國外文獻報道了位于外顯子86上的甘氨酸替代突變,分別是c.6797G>T(p.Gly2266Val)和c.6752G>A(p.G2251E),前者純合突變來自一個索馬里家庭[5],后者雜合突變來自一個日本家庭[6]。經搜索PubMed和萬方數據庫,國外報道并進行基因檢測的DDEB共54例(表1),其中44例為甘氨酸替代突變,并且44例甘氨酸替代突變中以位于外顯子73最多(23例),外顯子87次之(4例)。因此國外常見的DDEB點突變位于外顯子73。國內報道并進行基因檢測的DDEB共10例(表2),其中9例為甘氨酸替代突變,而9例甘氨酸替代突變中有4例位于73號外顯子。國內外DDEB以位于COL7A1基因73號外顯子的點突變最常見,本例突變位于86號外顯子,國內尚未見報道。

表1 國外報道的DDEB突變位點

續表

表2 國內報道的DDEB突變位點
綜上,我們報道來自廣東-DEB家族的COL7AI基因外顯子86號上的雜合突變:c.6761G>A;p.Gly2254Glu,這一點突變是國內外首次報道。
