COL1A2基因剪接突變所致成骨不全1例分析
2022-07-13 11:34:00王天平胡曼云貴州省貴陽市第四人民醫院內分泌科550005
醫學理論與實踐 2022年13期
關鍵詞:基因突變
王天平 胡曼云 張 喜 貴州省貴陽市第四人民醫院內分泌科 550005
成骨不全癥(Osteogenesis imperfecta,OI)或稱為脆骨病,是常見的單基因突變所致的遺傳性骨病,遺傳方式呈常染色體顯性遺傳(Autosomal dominant,AD)和常染色體隱性遺傳(Autosomal recessive,AR),以AD多見,但也有不少散發病例的報道。臨床上以骨量低下、骨骼脆性增加而反復發生骨折、骨骼畸形為主要特征,可伴有藍鞏膜、牙本質發育不良、聽力障礙、皮膚及韌帶松弛等骨骼外表現。主要涉及的致病基因是骨基質Ⅰ型膠原蛋白編碼基因及其代謝相關基因[1-2]。
1 病例資料
患兒男性,12歲,漢族,11歲時因爬樓踩滑(約于站立高度)致右側肩部及右膝關節骨折,外院行手術治療,隨后逐漸出現腰背部持續性隱痛,疼痛能耐受,行走時加重,日常活動(慢走、短距離爬樓)無明顯受限,但跑、跳等運動完成困難,上述癥狀持續約1年后再次就診外院,查維生素D:25-羥基維生素D2+D316.8ng/ml。腰椎片:椎體骨骺板骨軟骨炎可能。遂轉診于我院骨科門診查胸、腰椎MRI(見圖1):脊柱稍向后凸,胸腰段椎體變扁及信號增高原因,性質:骨質疏松可能。隨后以“成骨不全?”收入院。入院后詳細詢問病史,患兒出生時鞏膜就為藍色,約1歲時開始口服“魚肝油等”藥物,堅持飲牛奶200~400ml/d,持續2~3年后上述行為調整為間斷進行。患兒日常活動能力與其他同齡兒無明顯差別。患兒飲食上喜素食,近2年體重增加10+kg,身高增高約3cm。此次主要因為腰背部持續性疼痛就診欲查明原因。
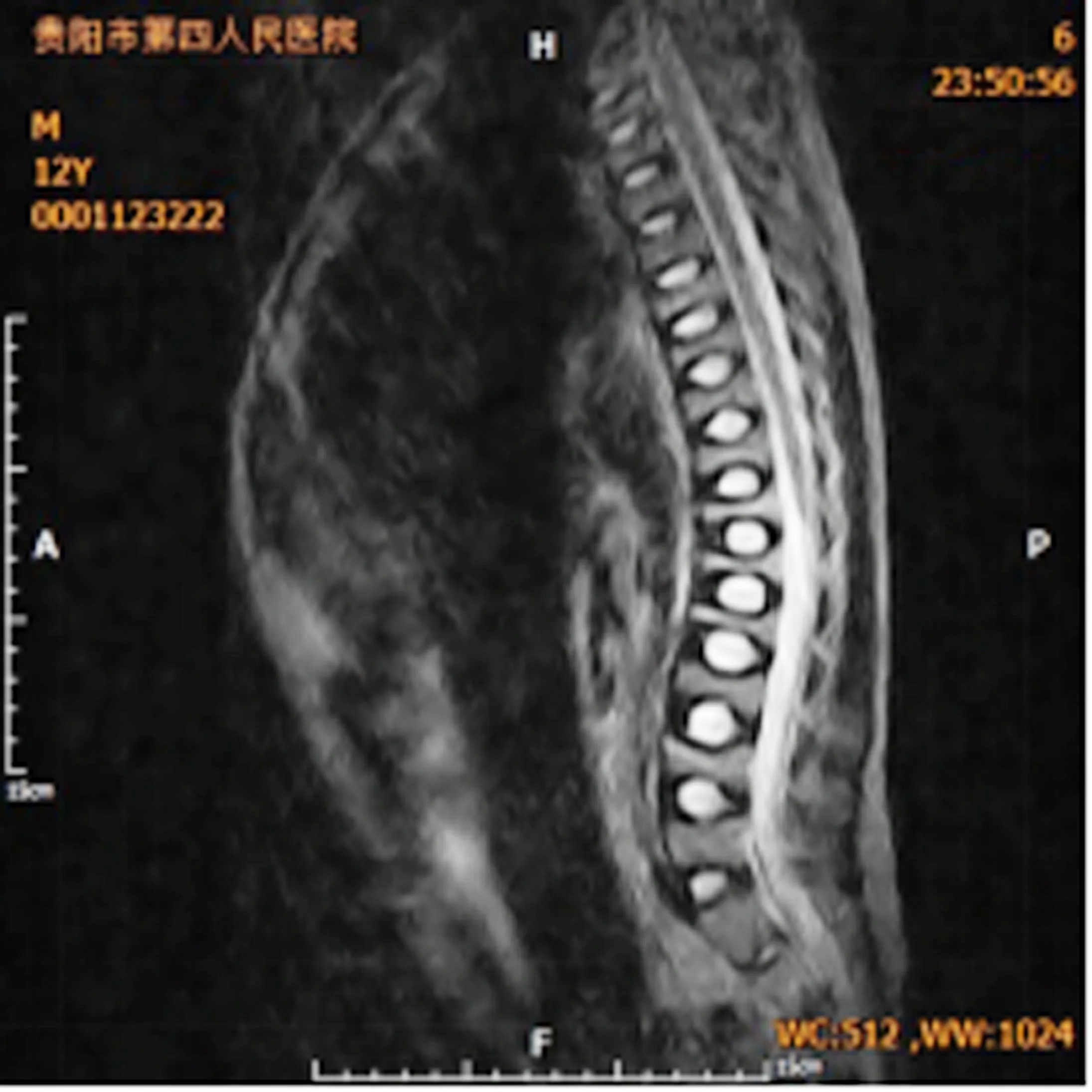
圖1 腰椎MRI
患兒母親孕期未產檢,患兒囟門5~6歲才完全閉合,時間較同齡兒明顯滯后,其他生長發育方面患兒父母未發現明顯異常。目前還未出現遺精等青春期啟動表現。9歲時因“左側小腦少突星形角質細胞瘤并出血性卒中”于外院手術治療。患兒直系親屬三代中均無身高異常矮、骨骼明顯畸形及脆性骨折情況。
體格檢查:生命體征平穩,身高:133cm,體重:35kg,指間距:137cm,上部量:73cm,下部量:60cm,坐高:70cm。雙眼鞏膜呈藍色。牙列稀疏。雙側腹股溝觸診未捫及包塊。雙側髂骨于腹部中部外側可捫及。脊柱胸腰段后凸畸形。雙手指關節彎曲畸形(見圖2),未見多指、趾,四肢指、趾關節均較柔軟(見圖3)。雙下肢稍呈“X”形表現,行走稍異常。余查體未見明顯異常。

圖2 雙手正面照片
輔助檢查及分析:(1)骨代謝相關的指標:PINP>1 200.00ng/ml,CTX 0.897ng/ml,甲狀旁腺激素 42.20pg/ml,骨源性堿性磷酸酶126.85μg/L,其中反映骨生成的指標PINP明顯增高,考慮為骨折后骨生成明顯增加有關。(2)胸腰椎正側位+骨盆+雙手正位片:①符合成骨不全X線征象;②雙手未見明顯異常。閱片可見骨質稀疏,骨小梁稀少。(3)染色體檢查提示46 XY,正常男性染色體表現。性激素未見異常。陰囊B超提示雙側睪丸存在。結合腹股溝查體情況不考慮性功能不全所致的繼發性骨質疏松。(4)皮質醇晝夜節律試驗提示皮質醇呈晝夜節律變化。結合查體情況不考慮皮質醇增多癥所致的繼發性骨質疏松。(5)五官科會診診斷考慮右耳傳導性聾(輕度)、脆骨—藍鞏膜—耳聾綜合征。其他輔查未見明顯異常。
治療:本例患兒前來我院就診時已經出現了右側肩部、右膝關節骨折及胸腰椎的多發壓縮性骨折,生活方式指導患兒提高肌肉強度及改善機體協調能力,注意避免頻繁跌倒所致骨折發生,并進行補鈣及補充維生素D3,因患兒家屬拒絕,住院期間沒有使用雙磷酸鹽抑制骨吸收治療,在之后的隨訪中,患兒再次發生了下肢的骨折,就診于外院手術治療骨折后,使用了雙磷酸鹽藥物抑制骨吸收。建議患兒的父母在下次備孕前進行有效的遺傳咨詢和懷孕后產前診斷,這有利于OI家庭的優生優育。
2 基因檢查及家系驗證
基因檢查結果:患兒存在COL1A2基因c.693+1G>C剪接突變(Het)。使用Sanger測序對家系成員驗證該突變位點,結果提示未見其他家系成員存在相同突變。Sanger測序圖譜見圖4。
患兒存在COL1A2基因c.693+1G>C剪接突變,為雜合突變;患兒父親、患兒母親、患兒大妹、患兒二妹測序結果未見相同突變
3 討論
患兒為1例攜帶COL1A2基因c.693+1G>C剪接突變(Het)的散發OI病例。根據患兒疾病的嚴重程度及臨床特征,考慮患兒OI Ⅰ型(Sillence分型)。患兒突出的臨床表現是反復發生脆性骨折,伴有藍鞏膜、聽力障礙、皮膚及韌帶松弛體征,基因檢查示COL1A2基因c.693+1G>C剪接突變(Het),Sanger測序行家系驗證未見其他家系成員存在相同突變。趙秀麗等[3]已通過研究構建了中國人OI相關的COL1A1/2基因突變譜,本例患者存在的COL1A2基因c.693+1G>C剪接突變在其突變譜中是可以查詢到的,證實該突變在中國人群中是OI的致病突變。
Ⅰ型膠原蛋白是骨有機質中含量最豐富的蛋白質,其代謝異常是OI發病的重要環節。Ⅰ型膠原蛋白分子由2條α1鏈和1條α2鏈構成穩定的異源三螺旋結構,α1鏈和α2鏈分別由COL1A1和COL1A2基因編碼,若COL1A1或COL1A2基因發生突變,其翻譯的前α1鏈或前α2鏈將形成異常三螺旋結構的前Ⅰ型膠原蛋白,從而導致Ⅰ型膠原蛋白合成的數量減少或蛋白功能、結構異常[4]。導致OI的致病基因有很多,其中以COL1A1、COL1A2兩種基因突變最多見,文獻報道占80%~90%[5],而攜帶COL1A1/2基因致病突變的OI患者中,COL1A1基因突變的檢出率會更高,目前報道的COL1A1基因突變有2 077種,COL1A2基因突變有1 089種(https://oi.gene.le.ac.uk,數據最后更新時間分別為2020年11月18日、2020年11月19日)。攜帶COL1A1/2基因突變的OI患者中,COL1A1基因突變的患者病情也會更嚴重,這與COL1A1、COL1A2基因分別編碼的α1鏈、α2鏈在Ⅰ型膠原蛋白分子中的不同作用相關。COL1A1/2基因突變的類型有錯義突變、剪接突變、插入和缺失、移碼突變等,其中最常見的是錯義突變[6],本例患兒存在的剪接突變并不是OI相關的COL1A1/2基因突變類型中最少見的,國內的兩項研究,分別對56例OI患者[7]和200例OI先證者及其家系成員[3]進行COL1A1/2基因突變鑒定,剪接突變的占比分別為17.9%、15.2%,均大于缺失或移碼突變等。
關于OI的疾病分型,1979年Sillence等將OI分為4型,主要根據患者的臨床表現及病情嚴重程度來劃分(傳統分型):其中Ⅰ型最輕微,無肢體變形;Ⅱ型圍產期死亡;Ⅲ型存在嚴重長骨畸形;Ⅳ型病情嚴重程度介于Ⅰ和Ⅲ之間,臨床表現不一。國際骨骼人類遺傳學疾病命名組織(INCDS)于2009年在Sillence分型的基礎上增加了1型,將OI分為5型,主要分型依據也是根據臨床表型和疾病嚴重度。這兩種疾病分型方法是大多數文獻中所提及的分型方法,這樣的臨床分型可以在不做基因檢測的情況下迅速判斷OI患者疾病的嚴重程度和臨床表型特征。但OI涉及的致病基因很多,突變類型也多種多樣,遺傳外顯性差異也較大,相同基因突變可能導致不同表現型,即使在同一家系也可能出現不同的表現型,目前國內外均已有針對很多疾病的基因靶向治療,對于OI患者是否能進行這方面的治療,有待更多學者的研究。那OI是否能根據致病基因對疾病進行分型以指導未來可能會有的基因靶向治療呢?Fratzl-Zelman則在分子遺傳學基礎上,主要根據OI致病基因在疾病病理生理過程中的生化作用將OI分成了11型(Ⅰ~Ⅺ),還有部分致病基因未分型,其中Ⅰ~Ⅳ型的具體分型方法基本同傳統分型方法。國內的學者[8]也提出了新的分型,根據患者的致病基因、遺傳方式,將OI分成了15型,仍有部分致病基因未分型。可見,隨著基因檢測技術在臨床上的廣泛使用,以致病基因為主要的分型依據已有趨勢。
COL1A2基因位于7q21.3,由52個外顯子組成,COL1A2基因正常表達的第一步就是要以DNA為模板轉錄形成mRNA,初始轉錄產物形成的是mRNA前體,需要經過加工和修飾,才能形成有功能的mRNA,這個過程中有一個重要環節就是要在剪接酶的作用下將內含子去除,當基因突變出現在內含子并導致剪接位點發生了改變后,就會造成mRNA前體的異常剪接,進而編碼出異常的前α2鏈,最終導致Ⅰ型膠原蛋白結構或功能異常。本例患兒攜帶COL1A2基因c.693+1G>C剪接突變(Het),該突變是發生在14號內含子,第693號核苷酸向后數1位的內含子核苷酸由G替換成了C,該突變就會導致mRNA前體的異常剪接,是已被報道的致病突變。進一步查詢相關的突變數據庫(https://databases.lovd.nl/shared/genes/COL1A2,訪問時間2021年12月7日),c.693+1這個位點的突變已報道了5例,國內外均有報道,DNA改變均為堿基替換,疾病診斷均考慮OI Ⅰ型,說明該突變位點及DNA突變方式并不是中國人群特有的,該突變方式所致的疾病分型上中國人群并無明顯異質性,當然這還需要更多數據的支持。
隨著臨床上基因檢測技術的廣泛使用,關于OI致病基因的文獻報告層出不窮,的確豐富了OI致病基因的突變譜,但目前仍無針對OI致病基因的相關治療,目前基本為對癥治療。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
