二例遺傳性大皰性表皮松解癥基因突變研究
2022-07-15 07:42:54廖曉捷于越乾王真真孫樂樂張福仁
中國麻風皮膚病雜志 2022年9期
關鍵詞:基因突變
廖曉捷 于越乾 王真真 孫樂樂 劉 紅 張福仁
山東第一醫科大學附屬皮膚病醫院(山東省皮膚病醫院),山東省皮膚病性病防治研究所,山東濟南,250022

遺傳性大皰性表皮松解癥(epidermolysis bullosa,EB)是一種罕見且嚴重的機械性皮膚病,在新生兒中的發病率約為19/106、患病率約為11/106,患者以皮膚或黏膜脆性增加、摩擦或創傷后水皰、大皰的形成為主要臨床特征[1]。我們對2例EB患者進行調查,應用全外顯子組測序技術進行分析,再用Sanger測序驗證結果,為臨床診斷提供確切的依據。
1 資料與方法
1.1 臨床資料 山東第一醫科大學附屬皮膚病醫院門診收集2例EB患者。散發患者1:女,19歲。四肢出現淡粉紅色或褐色的丘疹、結節3年,伴劇烈瘙癢,表面破潰,部分皮損愈合后可見結痂和萎縮性瘢痕。患者無類似疾病家族史,其父母非近親結婚。皮膚科檢查:雙前臂、雙脛前和雙足背可見大小不等的淡粉紅色或褐色的丘疹、結節和結痂,未見明顯水皰,無血皰(圖1a);雙足第一趾甲增厚萎縮(圖1b),口腔黏膜未受累。皮膚組織病理示:表皮角化過度、灶性角化不全,表皮下裂隙,真皮淺層血管、纖維組織增生,血管周圍輕度單一核細胞浸潤,符合大皰性表皮松解癥。結合患者的臨床表現和皮膚組織病理檢查,臨床診斷為EB。
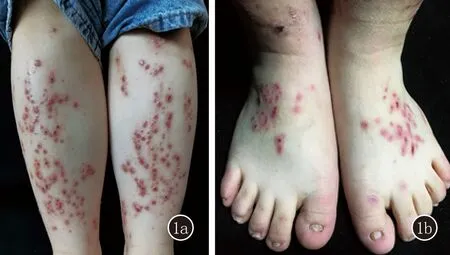
圖1 散發患者1臨床照片 雙脛前紫紅色結節、丘疹,部分結痂(1a),雙足第一趾甲增厚萎縮、營養不良(1b)
家系患者2:先證者,男,1歲。自出生雙手足背出現白色粟丘疹、水皰,伴右手中指指甲脫落(圖2a)。先證者足月生產,一般情況好,父母非近親婚配。皮膚科檢查:雙手足背白色粟丘疹、水皰伴皮膚破損,右手中指指甲脫落,無自覺癥狀,無血皰,手掌、肘部、膝部等摩擦部位和口腔黏膜均未受累。其父有類似病史,自幼雙手足背水皰,摩擦后加重,隨年齡增長水皰逐漸減輕,但出現雙脛前粟丘疹,伴劇烈瘙癢,可見糜爛結痂,愈合后留有白色萎縮性瘢痕;雙手足指趾甲均變薄、渾濁,遠端缺損(圖2b),未曾出現過手掌、足趾等部位的緊張性大皰。結合病史及皮損表現,臨床擬診為EB。先證者幼小,其父母拒絕對其行皮膚組織病理檢查和抽血檢查,為進一步明確診斷,對患兒父親進行全基因組外顯子測序。
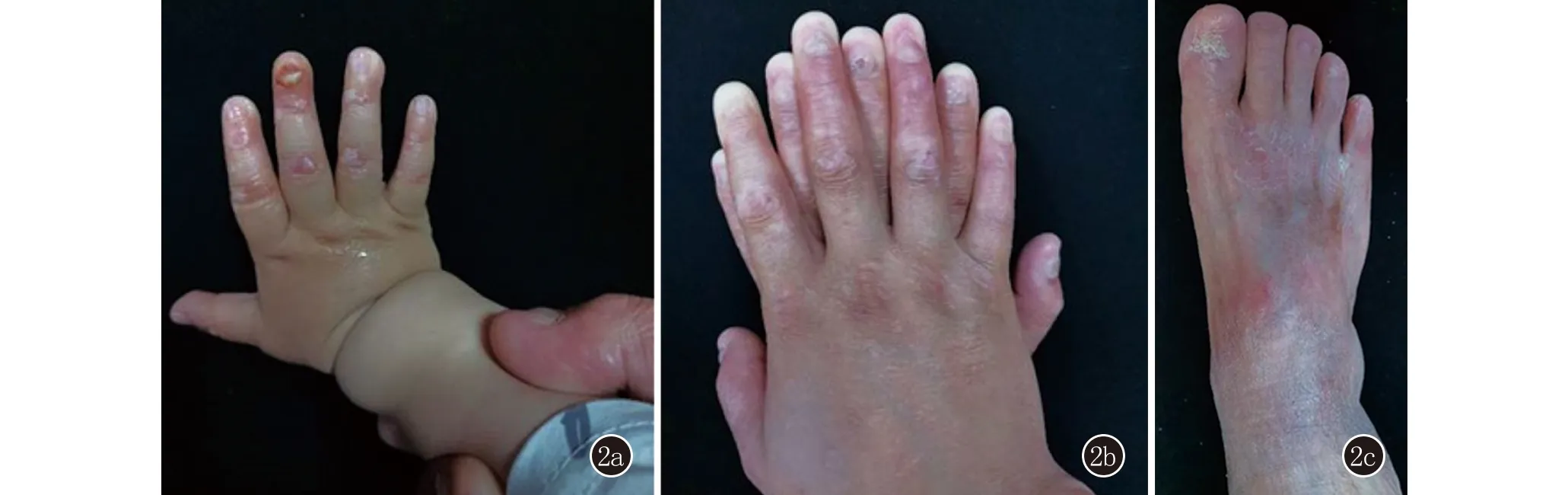
2a:先證者雙手白色粟丘疹、水皰,伴右手中指指甲脫落;2b,2c:其父雙手足白色粟丘疹,雙手足指趾甲均變薄、渾濁,遠端缺損
1.2 研究方法
1.2.1 樣本采集 經本院倫理委員會批準,經過家系成員的知情同意并簽字后,采集患者及其父母的外周血5 mL,EDTA抗凝,采用全自動核酸提取儀提取所有標本DNA,使用核酸濃度測定儀NanoDrop2000對標本 DNA的含量及純度進行測定,并將其標化為30 ng/μL的DNA模板。
1.2.2 變異篩選 將散發患者1、家系2先證者父親的外周血 DNA進行全基因組外顯子測序,首先使用AIExome Enrichment Kit V4試劑盒進行捕獲實驗和構建文庫,基因組文庫經過質量檢測合格后通過 Illumina HiSeq 6000 平臺完成高通量測序。測序原始數據采用BWA 軟件與參考基因組(hg19)進行比對,比對好的數據用GATK軟件進行變異位點的尋找,尋找的位點均通過Annovar軟件評估變異的影響并進行注釋。
1.2.3 Sanger測序驗證 根據全基因組外顯子測序篩選出的致病基因突變位點,通過NCBI數據庫查取突變基因的外顯子序列,應用Primer5.0軟件設計引物,對收集的外周血DNA樣本進行突變位點的Sanger測序驗證。PCR反應條件: 預變性94℃ 10 min、變形 94℃ 30 s、退火55℃ 45 s、延伸 72℃ 10 min,共35個循環之后72℃延伸10 min;取PCR反應產物經瓊脂糖凝膠電泳,將目的片段切膠純化后用ABI 3500Xl Dx Genetic Analyzer測序儀進行檢測。
1.2.4 突變分析 所有測序結果經NCBI blast數據庫、Chromas軟件與相應的突變基因參考序列進行分析比對,檢索Pubmed和萬方數據庫至今發表的關于本研究中突變位點的文章。
2 結果
2.1 臨床調查 調查患者的臨床資料,其中類似患者2例,分別為散發患者1和家系2先證者的父親,臨床癥狀均相似:均表現為雙脛前癢疹樣改變,伴有輕度甲營養不良;但發病年齡不同,前者是近成年才出現皮損,后者是出生后即有皮損,且水皰發生于四肢伸側,尤其是趾指關節面,愈后留有白色丘疹狀瘢痕,隨年齡增長,水皰性皮損逐漸減少,而出現癢疹樣損害。其余家屬均無相關臨床表現。
2.2 基因變異分析結果 散發患者1的COL7A1基因73號外顯子存在c.6082G>A雜合,導致編碼蛋白質的第2028位氨基酸由甘氨酸變成精氨酸(p.G2028R),其父母均未攜帶該突變(圖3)。該突變已在顯性營養不良型EB中報道過[2],屬于致病突變。
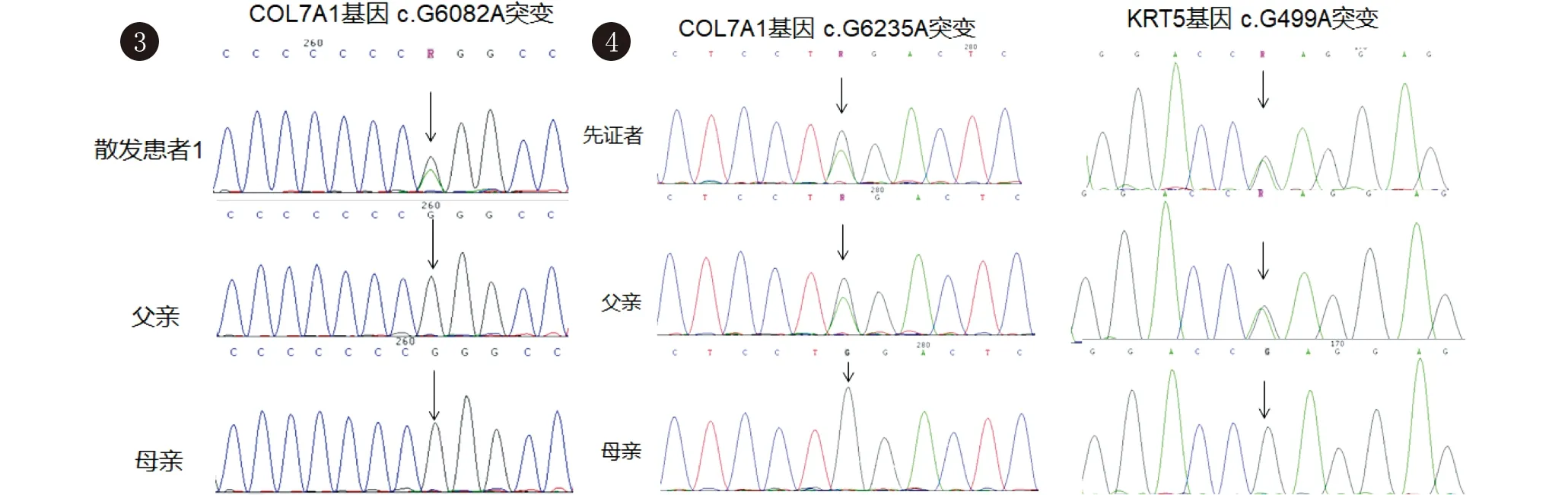
圖3 散發患者1及其父母測序結果 散發患者1攜帶COL7A1基因c.6082G>A突變,其父母均未攜帶此突變 圖4 家系2先證者及其父母測序結果 先證者及其父親同時攜帶COL7A1基因c.6235G>A突變和KRT5基因c.499G>A突變,母親未攜帶這2個基因突變
家系2先證者父親的全外顯子組測序結果顯示雜合2個基因的錯義突變,即COL7A1基因c.6235G>A(p.G2079R)突變和KRT5基因 c.499G>A(p.E167K)突變,經Sanger測序先證者也攜帶這2個突變,其母親無這些突變(圖4),證實先證者這2個基因的突變均來自其父親。本研究中COL7A1基因和KRT5基因的錯義突變分別在顯性營養不良型EB[3]和單純型EBS[4]中報道過,均屬于致病突變。
所有測序結果均通過反向測序驗證,且在與醫院自有的364個正常對照的全外顯子組測序數據中不存在這些突變。
3 討論
EB根據超微結構下水皰在皮膚基底膜帶發生的位置不同,最新的分類將其分為單純型EB(EBS)、交界型EB(JEB)、營養不良型EB(DEB)和kindle EB[5]。DEB的臨床特征為反復發生的水皰、癢疹樣結節、粟丘疹和萎縮性瘢痕,其水皰位于真皮基底膜帶致密板以下,位置深在,所以愈合后留有明顯的瘢痕[6]。
DEB均由編碼Ⅶ型膠原的COL7A1基因突變所致,本研究中2例患者的COL7A1基因突變既往報道均導致顯性遺傳的癢疹樣營養不良型大皰性表皮松解癥(dystrophic epidermolysis bullosa pruriginosa,DEB-Pr)。李桐等[7]在2014年于國內首次報道DEB-Pr中COL7A1基因p.G2079R該突變位點,文中先證者的表型與家系2先證者父親的表型相同。DEB-Pr于1994年首次由McGrath等[8]提出,其特征是好發于四肢伸側的癢疹樣結節、苔蘚樣斑塊及紫羅蘭色線狀瘢痕,伴劇烈瘙癢和指趾甲損害[9]。DEB-Pr常于嬰兒早期或兒童期發病,愈后留有白色丘疹樣瘢痕,伴粟丘疹形成和甲營養不良;少數病例于成年后發病,最晚有71歲發病的報道[10]。
COL7A1基因定位于在3p21,由118個外顯子組成,編碼Ⅶ型膠原蛋白,由Ⅶ型膠原構成的錨原纖維是基底膜的重要組成部分[11]。Ⅶ型膠原包括中央三螺旋桿狀膠原區域以及氨基端和羧基端,其中三螺旋區的甘氨酸替代的錯義突變為DEBP最常見的基因突變類型,Ⅶ型膠原蛋白結構特征是Gly-X-Y的連續重復[12]。甘氨酸是最小的氨基酸且位于三螺旋區的中央軸,此位置的蛋白鏈緊密折疊,只能容納最小的甘氨酸,所以甘氨酸殘基的替換會扭曲三螺旋的構象,膠原合成障礙后使錨原纖維數量或結構上發生異常,進而真表皮處的連接功能減弱、皮膚脆性增加,致密板下水皰形成[13]。本研究中2例患者的COL7A1基因突變位點p.G2028R和p.G2079R為發生在不同外顯子上的相同堿基替換,均為鳥嘌呤變成腺嘌呤,且均導致編碼的氨基酸由甘氨酸變成精氨酸,屬于經典的COL7A1三螺旋區內的甘氨酸置換,但發病時間完全不同,臨床表型和發病時間不僅取決于致病突變的性質,還受細胞類型、微環境和外部因素的影響[14]。COL7A1基因型和表型的關系尚不明確,該基因同一突變位點亦有較大的臨床異質性。Nakamura等[14]總結分析3例COL7A1基因中p.G2028R甘氨酸替代突變導致DEB,其表型有單純的甲營養不良、典型的DDEB表型和DEB-Pr的表型,首次證實相同的COL7A1基因甘氨酸替換可引起明顯的表型異質性。
角蛋白KRT5由基底層角質形成細胞表達,其中間絲蛋白基因突變會引起細胞骨架結構不穩定[15]。家系2中KRT5基因突變既往報道導致局限性EBS,表現為局限于手足部位反復發作的表皮內大皰,不累及黏膜和指甲,且愈后不留瘢痕[5],但家系2的先證者及其父親從未出現過手足等部位的緊張性大皰,無既往報道的相關臨床表型。角蛋白的結構從氨基端到羧基端分頭區、桿區和尾區三個部分,本研究中KRT5基因的錯義突變導致167位的酸性谷氨酸被堿性賴氨酸取代,突變位于角蛋白頭區H1的倒數第二個氨基酸,導致角蛋白絲組裝中斷,削弱角質形成細胞抵抗機械應力的能力而引起水皰,角蛋白不同結構區域的突變與EBS的嚴重性密切相關,發生于頭區H1的突變常引起輕微的臨床癥狀,可在超微結構上呈現出相對正常的角蛋白中間絲[16]。
家系2的先證者及其父親同時攜帶2個基因的錯義突變:COL7A1基因的c.6235G>A和KRT5基因的c.499G>A,雜合這2個基因的錯義突變尚未見報道。家系2未行透射電鏡、免疫熒光等檢測來定位裂隙水平,因此究竟是兩個基因突變還是單個基因突變發揮作用、家系2是DEB還是DEB和EBS兩亞型復合的EB類型均有待探究。 Vahidnezhad等[17]報道了兩基因共遺傳引起不同亞型共存的EB病例,先證者是EXPH5和COL17A1雙基因純合突變導致的伴發單純性和交界性的EB,在組織病理學和基因水平上同時具有EBS和JEB這2種不同類型的特點。本研究中先證者表現為手足白色粟丘疹、水皰及中指指甲脫落,尚無雙脛前癢疹樣結節,但其父表現出DDEB的臨床表型,家系2先證者隨訪半年未曾出現手掌和足趾等部位的大皰,并未出現EBS的臨床表型,這2個基因突變的共顯性遺傳并沒有加重每個基因單獨突變的后果,推測與KRT5基因該突變位點位于角蛋白頭區H1本身可導致十分輕微的臨床癥狀且EBS的皮損會隨年齡增長而改善有關。
綜上所述,2例患者經基因檢測的輔助診斷最終確診為EB,家系2雜合COL7A1基因和KRT5基因的錯義突變,同時攜帶這2個基因的雜合突變在國內外均為首報,基因檢測的結果拓展了EB的突變數據庫,但EB的臨床表型和基因型之間的關系仍有待于進一步研究。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
