雙重相對定量SD-qPCR在快速診斷性染色體變異中的應用
2022-07-29 12:45:20樊祖茜吳素萍林桂先凌秀明
檢驗醫(yī)學 2022年6期
關鍵詞:檢測
孫 雷, 龍 駒, 樊祖茜, 吳素萍, 林桂先, 凌秀明
(1.西安交通大學醫(yī)學部,陜西 西安 710061;2.欽州市婦幼保健院基因與遺傳實驗室,廣西 欽州 535099;3.欽州市婦幼保健院檢驗科,廣西 欽州 535099)
性染色體病又稱性染色體異常綜合征,是指由性染色體(X或Y)的數(shù)目異常或結(jié)構(gòu)畸變引起的疾病。性染色體病有多種類型,但都有共同的臨床特征,即性腺發(fā)育不全或兩性畸形。性染色體非整倍性是目前新生兒中最常見的染色體變異,總發(fā)病率為1/400,大約是唐氏綜合征發(fā)病率的2倍[1]。男性性染色體異常主要表現(xiàn)為47,XXY和47,XYY等異常,也可表現(xiàn)為48,XYYY、48,XXYY、48,XXXY或49,XXXXY等性染色體四體或五體異常,女性主要表現(xiàn)為性染色體單體45,X和性染色體三體47,XXX,也可表現(xiàn)為48,XXXX和49,XXXXX等X染色體四體或五體異常[2]。另外,還易出現(xiàn)1個或多個克隆的細胞失去或獲得性染色體,即發(fā)生性染色體嵌合體。最常見的性染色體嵌合體包括45,X/46,XX、46,XX/47,XXX、46,XY/47,XXY[3]。性染色體的異常率和異常種類非常多,因此對性染色體的產(chǎn)前診斷具有非常重要的臨床意義。
基于短串聯(lián)重復序列的熒光定量聚合酶鏈反應(short tandem repeat-quantitative fluorescent polymerase chain reaction,STR-QFPCR),多重連接探針擴增技術和熒光原位雜交(fluorescence in situ hybridization,F(xiàn)ISH)等技術可以實現(xiàn)非整倍體的檢測[4-8],但是由于需要特殊檢測儀器或操作相對繁瑣,不利于批量檢測。傳統(tǒng)的實時熒光定量聚合酶鏈反應(polymerase chain reaction,PCR)由于不需要擴增后的產(chǎn)物再處理步驟,在擴增的同時即可得到檢測結(jié)果,因此具有快速、簡便的特點。然而,由于傳統(tǒng)的熒光定量PCR采用的是不同的引物擴增不同的序列來進行相對定量,不同引物的擴增效率存在差異,從而導致檢測結(jié)果的穩(wěn)定性不足[9]。本課題組前期建立了基于重復序列的熒光定量聚合酶鏈反應(segmental duplication-quantitative polymerase chain reaction,SD-qPCR)方法,提高了對21號染色體診斷的穩(wěn)定性和可靠性[10]。據(jù)此,本課題組又開發(fā)出一種可進行雙重相對定量的SD-qPCR方法,以期能實現(xiàn)對其他染色體的快速檢測及單管多位點的檢測。
1 材料和方法
1.1 臨床樣本
收集197例羊水樣本,所有樣本經(jīng)染色體G顯帶核型分析,其中性染色體數(shù)目正常樣本185例、45,X樣本5例、47,XXX樣本3例、47,XXY樣本2例、47,XYY樣本2例。
1.2 重復序列的篩選
從美國國立生物技術信息中心(the National Center for Biotechnology Information,NCBI)數(shù)據(jù)庫(http://www.ncbi.org)中篩選出染色體間的重復序列分子標記,重復序列分子標記的一個位點必須位于性染色體(X或Y染色體),另一個位點位于性染色體以外的其他染色體。
1.3 引物和探針的設計
以篩選出的重復序列為目的序列,設計引物和探針。在重復序列堿基完全相同的部位設計公共擴增引物,使其能同時等效擴增重復序列的2個片段;在有堿基差異的部位設計各自的特異性檢測探針,使其能分別檢測出目的序列和參考序列的擴增量。
1.4 雙重相對定量SD-qPCR反應體系與參數(shù)
檢測儀器為CFX96實時熒光定量PCR儀(美國伯樂公司)。反應體系:總體積25 μL,模板DNA 2 μL、2對引物(5 μmol/L各0.5 μL)、引物對應的TaqMan探針(5 μmol/L各0.5 μL)、2×TaqMan Universal PCRMaster Mix[天根生化科技(北京)有限公司]12.5 μL、ddH2O 5.5 μL。熱循環(huán)條件:95 ℃ 10 min;95 ℃ 15 s,68 ℃1 min,循環(huán)5次,不收集熒光;然后95 ℃ 15 s,68 ℃ 1 min,于68 ℃時收集熒光,循環(huán)36次。
1.5 臨床樣本的檢測
采用建立的雙重相對定量SD-qPCR方法檢測197例羊水樣本,并根據(jù)16號染色體與X染色體間的重復序列2-ΔΔCt值來判斷X染色體的數(shù)目,1號染色體與Y染色體間的重復序列相對定量2-ΔΔCt值來判斷Y染色體的數(shù)目。將所有檢測結(jié)果與染色體核型分析結(jié)果進行對比,以判斷所建方法的準確性和可靠性。
2 結(jié)果
2.1 SD-qPCR引物和探針設計
經(jīng)N C B I數(shù)據(jù)庫篩選出了2對重復序列分子標記,定位于1 6號染色體(NC_000016.10)、X染色體(NC_000023.11)和1號染色體(NC_000001.11)、Y染色體(NC_000024.10),可分別用于檢測X和Y染色體的數(shù)目。SD-qPCR的引物設計原理及方法見圖1。
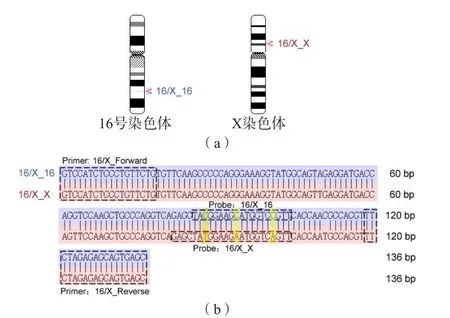
圖1 SD-qPCR引物設計原理及方法
2.2 SD-qPCR的濃度梯度檢測
采用SD-qPCR檢測不同濃度(160、32、6.4、1.28、0.256和0.128 ng/μL)的DNA,結(jié)果顯示,除DNA濃度為0.128 ng/μL時無擴增外,其他DNA濃度均能有效擴增,SD-qPCR的擴增曲線和標準曲線結(jié)果見圖2。
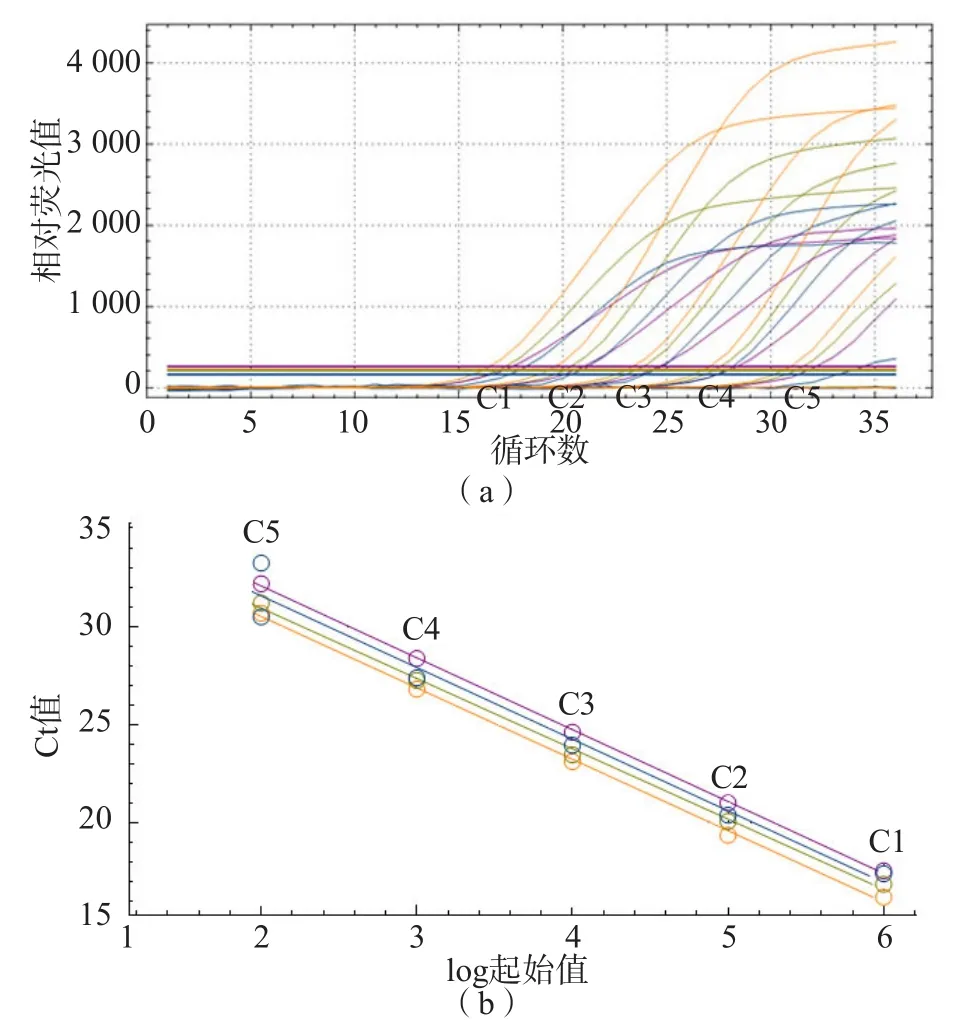
圖2 SD-qPCR的濃度梯度擴增曲線和標準曲線
計算不同DNA濃度的變異系數(shù)(coefficient of variation,CV)。對于16號染色體與X染色體間的重復序列,不同DNA模板濃度下ΔCt值的CV分別為6.37%、2.38%、5.65%、5.57%和8.82%。對于1號染色體與Y染色體間的重復序列,不同DNA模板濃度ΔCt值的CV分別為6.51%、3.47%、3.94%、6.73%和8.23%。
2.3 SD-qPCR檢測樣本的2-ΔΔCt值
性染色體拷貝數(shù)可根據(jù)16號染色體和X染色體、1號染色體和Y染色體的2-ΔΔCt值來判斷。樣本不同X和Y染色體數(shù)目的2-ΔΔCt值為:胎兒為女性或無Y染色體的樣本2-ΔΔCt(1-Y)值為0,有1條Y染色體的樣本2-ΔΔCt(1-Y)值為0.84~1.30,有2條Y染色體的樣本2-ΔΔCt(1-Y)值為1.90~2.01;胎兒為男性或僅1條X染色體的樣本2-ΔΔCt(16-X)值為0.37~0.65,有2條X染色體的樣本2-ΔΔCt(16-X)值為0.87~1.13,有3條X染色體的樣本2-ΔΔCt(16-X)值為1.34~1.45。各2-ΔΔCt值之間無相互重疊的情況,因此根據(jù)2-ΔΔCt值即可有效判斷X和Y染色體的數(shù)目。
2.4 臨床樣本的檢測結(jié)果
采用雙重相對定量SD-qPCR檢測197例羊水樣本,其中性染色體數(shù)目正常的樣本185例、45,X樣本5例、47,XXX樣本3例、47,XXY樣本2例、47,XYY樣本2例,檢測結(jié)果與染色體核型分析結(jié)果一致。不同染色體核型的雙重相對定量SD-qPCR檢測結(jié)果分布見圖3。
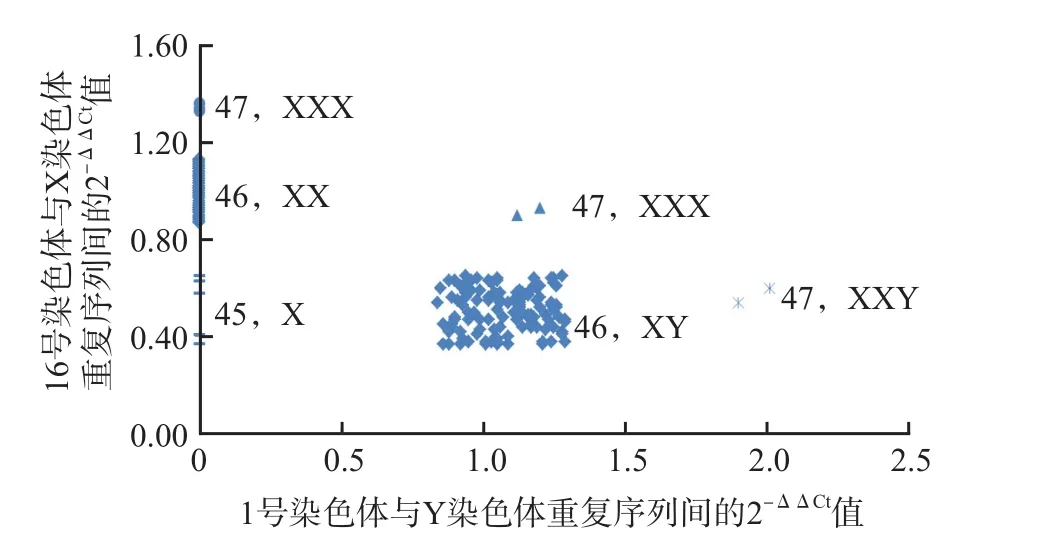
圖3 不同染色體核型的雙重相對定量SD-qPCR結(jié)果分布
3 討論
在人類基因組DNA中,不僅包含單一拷貝的DNA序列,還有大量的串聯(lián)重復序列(衛(wèi)星DNA、小衛(wèi)星DNA和微衛(wèi)星DNA)和分散重復序列。串聯(lián)重復序列中的衛(wèi)星和小衛(wèi)星DNA主要分布于著絲粒或端粒,常作為FISH技術的分子標記應用于遺傳疾病的診斷[5]。微衛(wèi)星DNA也被稱為短串聯(lián)重復序列(short tandem repeat,STR),可與定量PCR結(jié)合構(gòu)成STR-QF-PCR技術,用于常見染色體非整倍體的快速診斷[4]。分散重復序列是指重復單位并不相連,散布在整個基因組染色體各位點的序列,對其臨床應用方面的研究目前還較少。本研究采用的重復序列相當于只有2個拷貝的分散重復序列,是位于不同染色體上的2段具有堿基高度相似,但又有部分堿基數(shù)目或結(jié)構(gòu)差異的序列。
理想的重復序列分子標記應該是重復序列的兩端引物設計處堿基完全相同,而中間用于定量檢測的探針部分又有2個以上的堿基差異。這樣才能保證2個序列擴增效率的一致性,以及定量結(jié)果的特異性和準確性。通過多次的篩選和驗證,本研究成功篩選出2對重復序列分子標記,分別位于16號染色體、X染色體和1號染色體、Y染色體上。本研究采用2對引物和4條熒光探針同時擴增并檢測相應的染色體,從而開發(fā)了一種新的雙重相對定量SD-qPCR方法,雙重相對定量SD-qPCR的檢測結(jié)果與傳統(tǒng)的染色體核型分析結(jié)果一致,具有良好的準確性和穩(wěn)定性。
本研究采用雙重相對定量SD-qPCR對不同濃度梯度的DNA模板進行擴增,除在0.128 ng/μL濃度條件下無擴增外,DNA濃度為0.256~160 ng/μL時,所有的位點均能有效擴增,表明該技術的檢出限可達0.256 ng/μL。另外,本研究對各個濃度梯度的ΔCt值進行了CV分析,發(fā)現(xiàn)當DNA濃度為32 ng/μL時,16號染色體與X染色體間相對定量ΔCt值的CV為2.38%,1號染色體與Y染色體間的相對定量ΔCt值的CV為3.47%,該濃度的CV最小,表明該檢測體系的最佳模板濃度為32 ng/μL。
在前期研究中,我們主要通過比較待測樣本與正常樣本的ΔCt值差異來判斷待測樣本的基因或染色體的拷貝數(shù)[10]。通過ΔCt值區(qū)間的統(tǒng)計分析,可以有效檢出同一個批次待測樣本的染色體拷貝數(shù),具有簡便和直觀的特點。但當PCR儀器、試劑或DNA質(zhì)量有變化時,ΔCt值的波動會相對較大。因此,ΔCt值分析不適用于不同批次間的分析比較。鑒于此,本研究統(tǒng)一采用2-ΔΔCt值對樣本進行分析[11],Y染色體拷貝數(shù)可通過1號染色體和Y染色體重復序列的2-ΔΔCt(1-Y)值來判斷,X染色體拷貝數(shù)可通過16號染色體與X染色體重復序列的2-ΔΔCt(16-X)值來判斷,有效避免了采用ΔCt值分析的不足。
傳統(tǒng)的相對定量PCR雖然具有快速和簡便的特點,但存在穩(wěn)定性差和檢測位點少等缺點。(1)檢測的穩(wěn)定性:傳統(tǒng)的熒光定量PCR在基因表達量檢測方面應用較多[12-13],具有較好的準確性和重復性,原因主要是在科研實驗全過程中,人員、試劑和儀器等條件基本沒有變化,但當涉及不同的實驗室或人員、儀器等出現(xiàn)變動時,極其微小的差異就會導致擴增效率的改變,從而導致假陰性或假陽性結(jié)果[14]。(2)多位點的檢測能力:傳統(tǒng)的PCR檢測由于穩(wěn)定性不足,不同探針或引物間的干擾較大,一般只能進行單重相對定量PCR檢測[15],即單管只能檢測1條序列或染色體。本研究建立的雙重相對定量SD-qPCR方法使用的2對重復序列的擴增引物完全相同,從根本上解決了傳統(tǒng)PCR技術穩(wěn)定性不足的問題,并可以實現(xiàn)單管四色熒光雙重相對定量PCR檢測,單管即可同時檢測4條染色體。
綜上所述,建立的雙重相對定量SD-qPCR方法與傳統(tǒng)的PCR相比,具有檢測位點更多、檢測結(jié)果更穩(wěn)定的優(yōu)點。本方法不僅具有重要的臨床推廣和應用價值,同時也可為其他染色體數(shù)目異常的分子診斷方法的建立提供參考。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2019年9期)2019-11-25 07:34:36
中學生數(shù)理化·七年級數(shù)學人教版(2019年9期)2019-11-25 07:34:34
中學生數(shù)理化·七年級數(shù)學人教版(2019年12期)2019-05-21 02:53:50
中學生數(shù)理化·七年級數(shù)學人教版(2019年12期)2019-05-21 02:53:48
