
一測多評法測定雙黃連注射液中4 種成份的含量
2022-07-31 04:55:26王奎鵬徐興敏方清朝王均偉王祖紅
中國合理用藥探索 2022年1期
王奎鵬,徐興敏,方清朝,王均偉,王祖紅
1 河南中醫藥大學第一附屬醫院,鄭州 450000;
2 河南科技大學法醫學院,洛陽 471000;3 河南福森藥業有限公司,南陽 474450
雙黃連注射液由金銀花、黃芩和連翹按1∶1∶2的比例配伍,并經過提取精制而成。其具有清熱解毒、清宣風熱之功效,可治療外感風熱引起的發熱、咳嗽、咽痛,也適用于病毒及細菌感染所致的上呼吸道感染、肺炎、扁桃體炎、咽炎等,收載于原《中華人民共和國衛生部藥品標準中藥成方制劑》第18冊[1]。現行國家藥品標準WS3-B-2104-96-2010通過測定綠原酸、咖啡酸、黃芩苷和連翹苷4 個成份的含量,對雙黃連注射液進行質量評價。目前雙黃連注射液的含量測定均采用外標法(external standard method,EMS),即在相同的色譜條件下,以待測組份的純品為對照,比較對照品和樣品中待測組份的響應信號進行定量[2-6]。中藥成份復雜,僅檢測單一藥效成份無法作出客觀全面的評價,而多組份同時測定要求具備多個對照品,部分中藥對照品不易購置或制備,影響了中藥質量評價的研究進程[7]。一測多評法(quantitative analysis of multi-components by single marker,QAMS)基于多指標質量控制的研究思路,解決了參比物質價格高或制備困難等難題,可實現同時檢測多指標成份,已成為中藥及其制劑質量評價的新模式[8]。當前國內外尚無采用QAMS 測定雙黃連注射液成份含量的報道。基于此,本實驗以連翹苷為內標參照物,計算綠原酸、咖啡酸、黃芩苷的相對校正因子(fs/i),并測定各成份含量。采用EMS 同步測定,驗證QAMS 的準確性和可行性,為評價雙黃連注射液的質量提供參考依據。
1 材料
1.1 儀器
Agilent 1260 高效液相色譜儀[包括四元泵、VWD 檢測器、ChemStation 化學工作站,安捷倫科技(中國)科技有限公司];BP211D 電子分析天平(德國賽多利斯公司);YMC-C18 色譜柱(150mm×4.6mm,5μm,日本YMC 公司);KQ-700VDE 型雙頻數控超聲波清洗器(昆山市超聲儀器有限公司);YB-Z 型真空恒溫干燥箱(天津藥典標準儀器廠)。
1.2 試藥
綠原酸(批號:110753-201817,純度96.8%)、咖啡酸(批號:110885-201703,純度99.7%)、黃芩苷(批號:110715-201821,純度95.4%)、連翹苷(批號:110821-201816,純度95.1%)均購自中國食品藥品檢定研究院;乙腈(色譜純,美國霍尼韋爾公司);甲酸(色譜純,中國醫藥集團有限公司);超純水(河南福森藥業有限公司);雙黃連注射液(河南福森藥業有限公司,批號:202009061、202009071、202009081、202009091、202009101)。
2 方法與結果
2.1 色譜條件
采 用YMC-C18 色 譜 柱(150mm×4.6mm,5μm);以乙腈-0.2%甲酸為流動相進行梯度洗脫(見表1),程序結束后運行5min;檢測波長為324nm(綠原酸、咖啡酸)和280nm(黃芩苷、連翹苷);柱溫為35 ℃;進樣量10μl;流速為1.0ml/min。理論板數按連翹苷峰計算應不低于6000,綠原酸、咖啡酸、黃芩苷、連翹苷與相鄰色譜峰的分離度應達到1.5 以上。
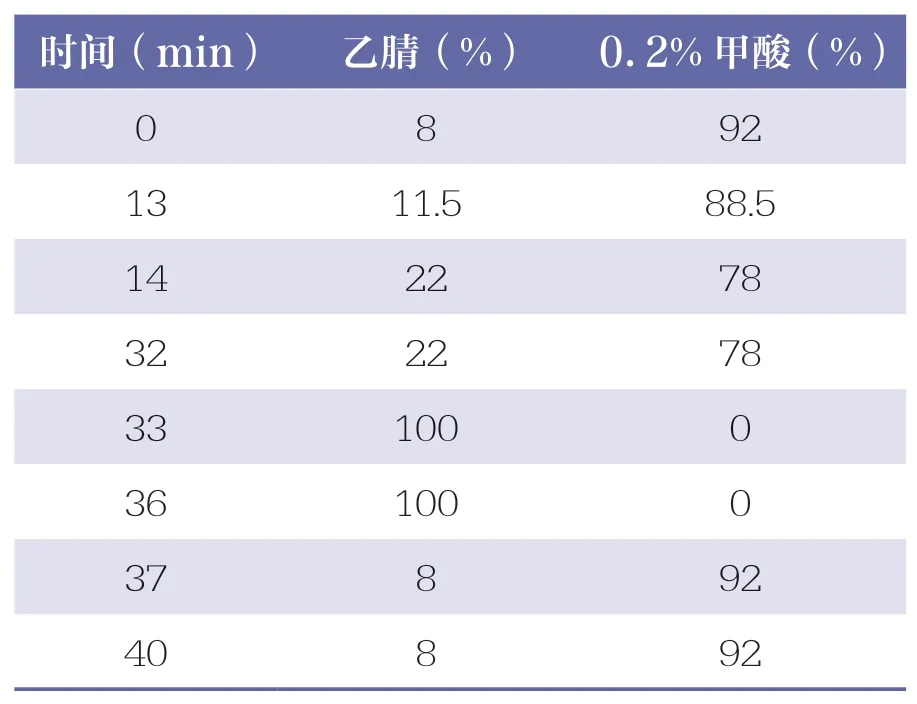
表1 洗脫梯度程序
2.2 溶液制備
2.2.1 混合對照品溶液的制備
精密稱取綠原酸、咖啡酸、黃芩苷和連翹苷對照品適量,置棕色量瓶中,加 50%甲醇制成質量濃度分別為40、30、60、60μg/ml 的混合對照品溶液,室溫保存,備用。
2.2.2 供試品溶液的制備
精密移取供試品2ml,置50ml 棕色量瓶中,加50%甲醇稀釋至刻度,搖勻,過濾,即得。
2.2.3 陰性對照品溶液的制備
按雙黃連注射液的生產工藝,制備無黃芩、金銀花、連翹的陰性樣品,按“2.2.2”項下方法制備陰性對照品溶液。
2.3 方法學考察
按“2.1”項下色譜條件,分別精密吸取混合對照品溶液、供試品溶液和陰性對照品溶液10μl,注入液相色譜儀,記錄色譜圖,見圖1。
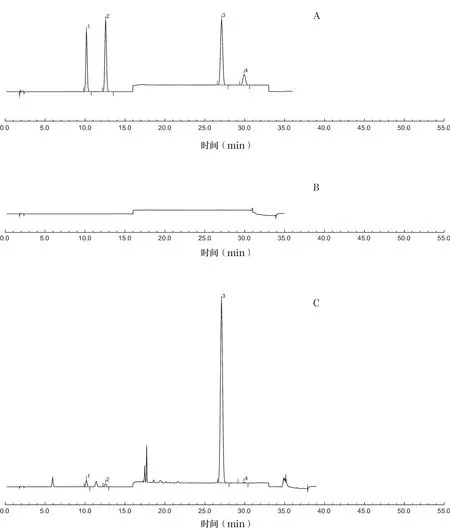
圖1 混合對照品(A)、陰性對照品(B)和樣品(C)的高效液相色譜(HPLC)圖
2.3.1 線性關系
分別精密吸取“2.2.1”項下混合對照品溶液1、3、5、10、15、20、30、50、80、100μl,按“2.1”項下色譜條件進行測定,記錄色譜圖。以進樣量(X)為橫坐標,峰面積(Y)為縱坐標進行線性回歸,詳見表2。結果表明,各成份在各自范圍內的線性關系均良好。
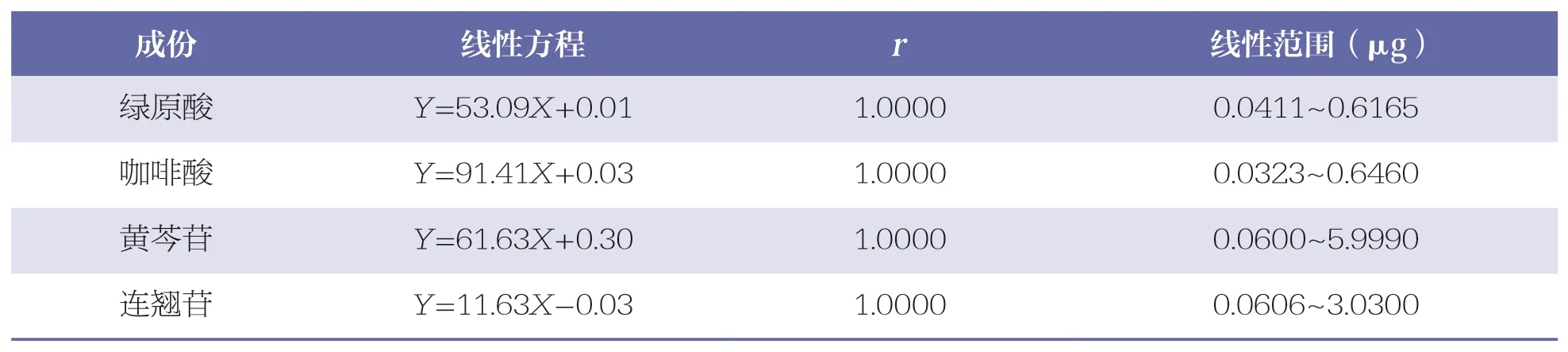
表2 4 種成份線性關系
2.3.2 精密度試驗
取同一供試品(批號:202009101),按“2.2.2”項下方法制備供試品溶液,按“2.1”項下色譜條件測定,連續進樣 6 次,測得綠原酸、咖啡酸、黃芩苷和連翹苷的峰面積RSD 分別為0.18%、0.43%、0.18%、1.67%,表明儀器精密度良好。詳見表3。
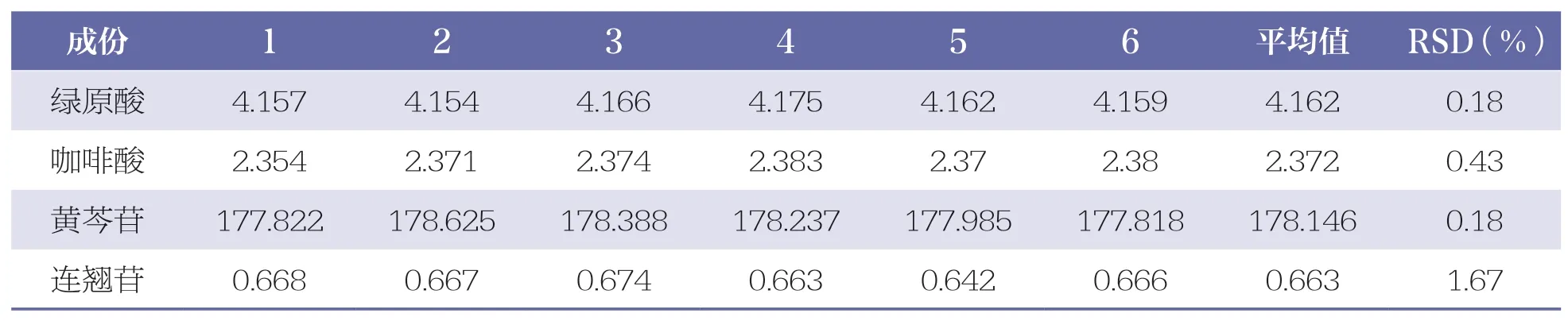
表3 4 種成份精密度試驗結果
2.3.3 重復性試驗
取同一供試品(批號:202009101)6 份,按“2.2.2”項下方法制備供試品溶液,按“2.1”項下色譜條件測定,記錄各色譜峰峰面積。綠原酸、咖啡酸、黃芩苷、連翹苷的平均含量分別為0.195、0.065、7.108、0.143 mg/ml,RSD 分別為0.21%、0.63%、0.21%、0.86%,表明該方法重復性良好。詳見表4。
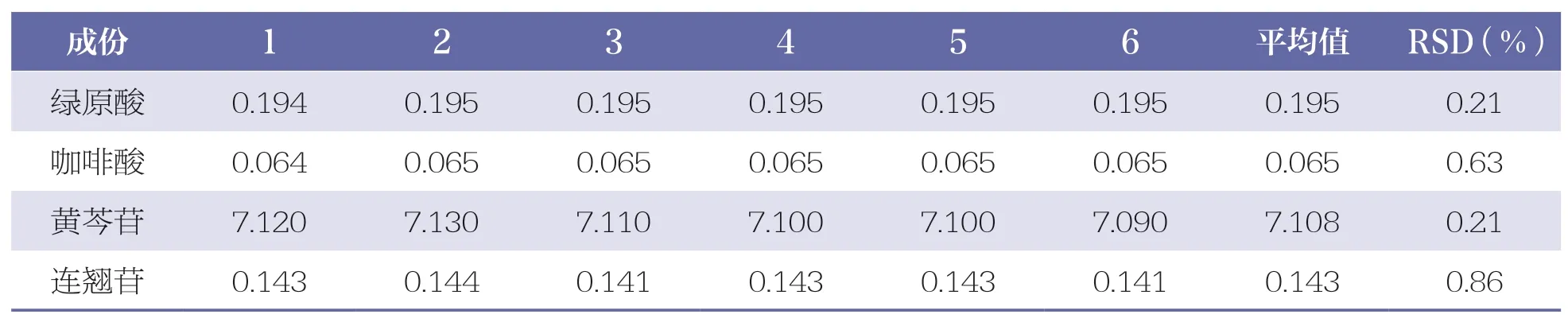
表4 4 種成份重復性試驗結果
2.3.4 穩定性試驗
取同一供試品(批號:202009101),按“2.2.2”項下方法制備供試品溶液,分別于0、2、4、6、8h進樣測定,測得綠原酸、咖啡酸、黃芩苷、連翹苷的RSD 分別為0.11%、0.29%、0.04%、1.42%,表明供試品溶液在8h 內穩定性良好。詳見表5。

表5 4 種成份的穩定性試驗結果
2.3.5 加樣回收率試驗
分別精密吸取含量已知的樣品溶液(批號:202009101)2.0ml,共6 份,分別加入各對照品溶液適量,按“2.2.2”項下方法制備供試品溶液,按“2.1”項下色譜條件測定含量,計算加樣回收率和RSD。綠原酸、咖啡酸、黃芩苷、連翹苷的平均加樣回收率分別為100.97%、100.08%、94.49%、101.10%,RSD 分別為0.26%、0.25%、0.32%、0.45%,表明測定結果準確。詳見表6。
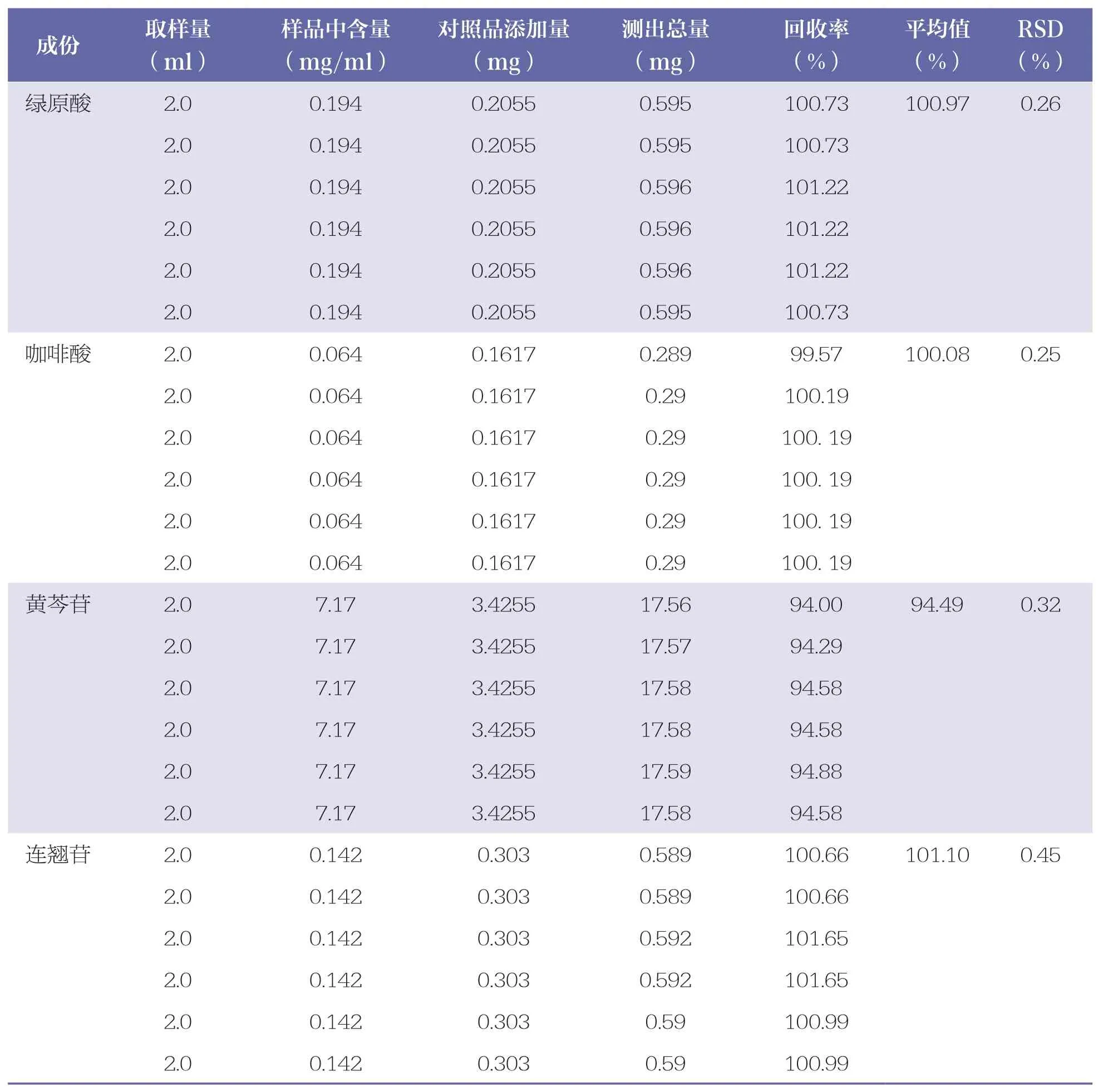
表6 4 種成份的加樣回收率試驗
2.4 相對校正因子(fs/i)的建立及耐用性評價
2.4.1 相對校正因子(fs/i)的建立
精密吸取“2.2.1”項下混合對照品溶液1、3、5、10、15μl,按“2.1”項下色譜條件進行測定,記錄各對照品中各組份的峰面積。以連翹苷為內標參照物,根據公式fs/i=(Wi×As)/(Ws×Ai),分別計算綠原酸、咖啡酸、黃芩苷的相對校正因子。式中,fs/i為相對校正因子;Ws為內標參照物的質量(μg);As為內標參照物的峰面積;Wi為待測成份的質量(μg);Ai為待測成份的峰面積。詳見表7。
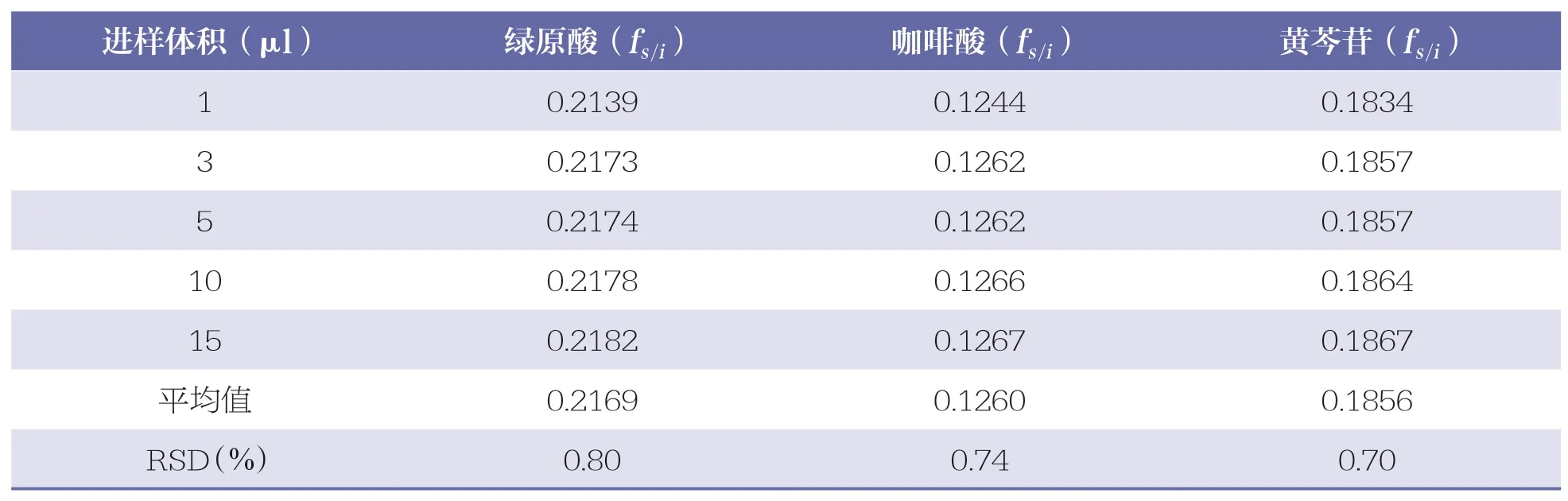
表7 樣品中待測成份的相對校正因子
2.4.2 相對校正因子(fs/i)的耐用性評價
考察不同高效液相色譜儀(Agilent 1260、賽默飛 DGP-3600SDN 和Waters E2489)對待測成份相對校正因子(fs/i)的影響。結果綠原酸、咖啡酸、黃芩苷的相對校正因子(fs/i)RSD 分別為 0.79%、0.76%、0.70%,表明不同色譜儀對3 種成份的相對校正因子(fs/i)無顯著影響。
考察不同型號色譜柱[Agilent Zorbax SBC18(150mm×4.6mm,5μm)、Syncronis C18(150mm×4.6mm,5μm)、YMC-C18(150mm×4.6mm,5μm)]對待測成份相對校正因子(fs/i)的影響。結果綠原酸、咖啡酸、黃芩苷的相對校正因子(fs/i)RSD 分別為1.20%、1.90%、1.50%,表明不同色譜柱對3 種成份的相對校正因子(fs/i)無顯著影響。
2.5 連翹苷對照品溶液的穩定性試驗
取連翹苷(批號:110821-201816,純度95.1%)對照品溶液,分 別于0、1、2、3、4、5、6、7個月進樣測定,測定連翹苷對照品的含量分別為99.98%、100.43%、101.58%、99.97%、98.96%、100.38%、100.30%、99.68%,RSD 為0.09%, 表明連翹苷對照品溶液在7 個月內穩定性良好。
2.6 樣品含量測定
精密吸取 5 個批次的雙黃連注射液供試品溶液各10μl,按“2.1”項下色譜條件進樣測定。采用EMS[9]和QAMS 分別計算供試品中各待測成份的含量,詳見表8。EMS 和QAMS 所計算出的結果無統計學差異(P>0.05),表明QAMS 可用于雙黃連注射液中綠原酸、咖啡酸、黃芩苷、連翹苷的定量分析。

表8 4 種成份的含量測定結果
3 討論
3.1 檢測波長的選擇
依據2020 年版《中國藥典》和紫外掃描測定結果可知,黃芩苷、連翹苷、綠原酸、咖啡酸分別在274、280、324、323nm 處有最大吸收,在同一波長處同時測定4 種成份具有難度。由于連翹苷的含量較低,且在324nm 處基本無吸收,因此確定連翹苷與黃芩苷的檢測波長為 280nm,并在檢測時增加進樣量[5];綠原酸類化合物和咖啡酸均含有咖啡酰結構,在 324nm 處有最大吸收,故設定綠原酸和咖啡酸的檢測波長為324nm[6]。綜合考慮各峰高及峰面積的比例,選擇324nm 和280nm 為最終檢測波長,在線進行波長切換檢測。
3.2 流動相和洗脫梯度的選擇
試驗中考察了乙腈-0.07%甲酸溶液[3]、0.6%冰醋酸水溶液-乙腈[10]、乙腈-0.2%甲酸[11]等不同流動相以不同比例進行成份洗脫,最終以乙腈-0.2%甲酸為流動相進行梯度洗脫。該條件下樣品分析時間短(40min 內即完成一針進樣檢測),柱效高,色譜圖基線平穩,峰形對稱,對照品出峰時間適中,主峰與其他峰的分離度、主峰的理論板數等色譜參數均符合2020 年版《中國藥典》的要求。
3.3 內標參照物的選擇
連翹苷不耐高溫,50℃以上會破壞連翹苷的結構使其變性,但在室溫條件下穩定性良好;溶液的酸堿度對連翹苷穩定性影響顯著,pH 為7 時最穩定;連翹苷的光穩定性好[12]。本試驗在7 個月內連續8次測定同批號的連翹苷對照品,結果表明其穩定性良好。
綜上,2015 年版《中國藥典》將QAMS 用于中藥提取物及中藥制劑的多指標質量控制。近年來在中藥材、中藥飲片及中藥制劑的質量控制中,QAMS 的應用范圍已由同類成份的測定擴展至多類成份的測定[13]。本研究結果表明,QAMS 具有簡便可行、準確、快速、重現性好、專屬性強的特點,可有效降低質控成本、提高質控效率,可用于雙黃連注射液的質量評價。
