絨毛崖豆藤中一種查爾酮提取物的體內外代謝及藥代動力學研究
2022-08-01 08:27:46倪恒凡
天然產物研究與開發 2022年7期
關鍵詞:血漿
倪恒凡,劉 玲,萬 麗*
1四川大學華西醫院藥劑科,成都 611137;2成都中醫藥大學藥學院,成都 610041
天然化合物對新藥的發現做出了巨大貢獻,黃酮類化合物是天然產物中的一大類[1],因為其具有良好的抗炎、抗腫瘤、抗菌、抗潰瘍、保肝、保護心血管等活性備受大家關注[2-9],從豆科崖豆藤屬植物絨毛崖豆藤(MillettiavelutinaDunn)莖中提取分離出的抗炎活性化合物2,5-二甲氧基呋喃[4″,5″:3,4]查耳酮(2,5-dimethoxyfuran[4″,5″:3,4]chalcone,1)屬于查爾酮類化合物。Ma等[10]研究結果顯示,該化合物具有較強的抗炎活性,其在體外細胞實驗中主要通過抑制IL-1β分泌和Caspase-1激活,并抑制ASC齊聚,從而表現出良好的抗炎活性。并在小鼠體內實驗中可明顯緩解LPS誘導的小鼠急性休克。在新藥開發的早期階段,進行藥物代謝的相關研究有助于快速識別安全、有效、藥代動力學性質良好的化合物,提前淘汰不合格的化合物,節約研發成本,目前尚未在國內外期刊上發現對該化合物的體內外代謝數據研究報道。本研究通過對2,5-二甲氧基呋喃[4″,5″:3,4]查耳酮在大鼠、小鼠、恒河猴、Beagle犬和人5個種屬肝微粒體和大鼠體內的代謝產物研究,并通過對2,5-二甲氧基呋喃[4″,5″:3,4]查耳酮及其代謝產物(M1)體內外藥代動力學研究探究該化合物的主要物質作用基礎,為其進一步的機制研究奠定基礎。
1 材料與方法
1.1 材料
美國Thermo公司Q Exactive高分辨質譜(含ESI源,軟件Xcalibur3.2、Compound Discover 2.0);美國AB SCIEX公司QTRAP5500三重四級桿質譜儀(軟件Analyst 1.6.2、MμLtiQuant 3.3);日本Shimadzu公司超高速液相色譜,美國Waters公司ACQUITY UPLC?BEH C18色譜柱。
樣品1(自制,No:20190221,純度≥98%,結構見圖1),ESI-MS:m/z309.122 1(calcd for C19H16O4,309.112 7)[M+H]+[10]。樣品M1(自制,No:20190311,純度≥98%,結構見圖1),ESI-MS:m/z311.1277(alcd for C19H18O4,311.1283)[M+H]+。內標辛二酰苯胺異羥肟酸(大連美侖生物技術有限公司,No:00502A,純度≥98%)。人種屬的肝微粒體(20 mg/mL)、大鼠種屬的肝微粒(20 mg/mL)、比格犬種屬的肝微粒體(20 mg/mL)、猴種屬的肝微粒體(20 mg/mL)、小鼠種屬的肝微粒體(20 mg/mL),NADPH反應啟動液(武漢普萊特生物醫藥技術有限公司),于-80 ℃冷凍保存。甲醇、乙腈(色譜純,Sigma-Aldrich公司)。甲酸(色譜純,FlukaAnalytical公司)。
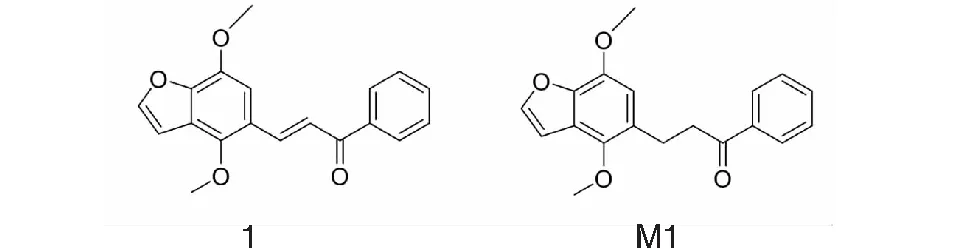
圖1 樣品1及M1結構式Fig.1 Structures of sample 1 and M1
實驗動物:SPF級SD雄性大鼠,體重200±10 g,SPF級C57雄性小鼠,體重22±2 g均購買于成都達碩實驗動物有限公司,許可證號:SCXK(川)2014-028。
1.2 溶液的配制
1.2.1 給藥溶液的配制
樣品1注射液制備方法:精密稱取樣品1適量,加入生理鹽水適量,制備成濃度1 mg/mL,于4 ℃冰箱密封保存,備用。
1.2.2 儲備液的配制
精密稱取樣品1及M1適量,用甲醇溶解制備成質量濃度為1 mg/mL的儲備液,于4 ℃冰箱密封保存,備用。
1.2.3 內標溶液的配制
精密稱取SAHA對照品適量,用乙腈稀釋成20 ng/mL的內標工作液。
1.3 LC-MS/MS條件
1.3.1 液相條件
QTRAP5500三重四級桿質譜:ACQUITY UPLC?BEH C18色譜柱(2.1 mm×50 mm,1.7 μm),流動相為:0.1%甲酸水(A相),乙腈(B相)梯度洗脫(0~1 min,10%→90% B,1~3 min,90% B),流速為0.5 mL/min,柱溫35 ℃,平衡1.5 min,進樣體積1 μL。
Q Exactive高分辨質譜:ACQUITY UPLC?BEH C18色譜柱(2.1 mm×50 mm,1.7 μm),流動相為:0.1%甲酸水(A相),乙腈(B相)梯度洗脫(0~15 min,10%→90% B,15~20 min,90% B),流速為0.5 mL/min,柱溫35 ℃,平衡1.5 min,進樣體積2 μL。
1.3.2 質譜條件
QTRAP5500三重四級桿質譜:采用MRM(多反應檢測模式)檢測,質譜條件為:ESI+(正離子模式),樣品1監測離子對為:m/z309.1→m/z267.1,碰撞能量為17.73 V;M1監測離子對為:m/z311.1→m/z191.1,碰撞能量為15.27 V;內標SAHA監測離子對為:m/z265.1→m/z232.1,碰撞能量為20 V;去簇電壓(DP)均為100 V;離子束聚焦電壓(EP):10 V;碰撞池出口電壓(CXP):10 V;駐留時間為0.10 s。
Q Exactive高分辨質譜:質譜條件為:ESI+(正離子模式),噴霧電壓(IS):4 500 V;霧化氣壓力(GS1):50 Psi;氣簾氣壓力(CUR):15 Psi;輔助氣壓力(GS2):45 Psi;離子源溫度(TEMP):550 ℃;簇裂解電壓(DP):55 V;碰撞能量(CE):35 V。
1.4 樣品1在各種屬代謝產物研究
配制孵育體系為200 μL(PBS 188 μL、NADPH 12 μL),再加入樣品12 μL(2 mmol/L),恒溫水浴預孵育(37 ℃、5 min),再加入各種屬的肝微粒體5 μL(20 mg/mL)啟動反應。在37 ℃水浴鍋中繼續孵育60 min后加入400 μL含SAHA(20 ng/mL)的冰乙腈終止反應。渦旋3 min,離心15 min(13 000 r/min),取上清液進樣,實驗平行3份[11]。樣品進UPLC-QE-Orbitrap-MS檢測,通過對比孵育0 min和60 min樣品離子流圖,尋找樣品1在各種屬肝微粒體中的代謝產物。
1.5 樣品1在C57小鼠體內代謝產物研究
隨機取C57小鼠分為對照組、血漿組和糞尿組,每組3只。將小鼠放置在代謝籠中,給藥前12 h禁食不禁水,對照組尾靜脈注射生理鹽水,實驗組注射樣品1(10 mg/kg)。血漿組分別于給藥后0.5、1.5 h收集血液樣品,尿液和糞便樣品于給藥后收集48 h內的糞便和尿液(每8 h收集一次)。取出離心轉速為3 500 r/min,時間15 min,后取上清液即得血漿樣品,樣品保存于低溫冰箱-80 °C。樣品預處理如下:
血漿樣品:取0.1 mL血漿樣品加入到預先活化的SPE小柱中,用1 mL 10%甲醇洗滌3次,棄去,再加入1 mL甲醇,將洗脫液進行氮吹,后加100 μL甲醇復溶,13 000 r/min離心15 min,取上層清液進樣分析。
尿液樣品:取0.5 mL尿液樣品加入到預先活化的SPE小柱中,用1 mL 10%甲醇洗滌3次,棄去,再加入1 mL甲醇,將洗脫液進行氮吹,后加100 μL甲醇復溶,13 000 r/min離心15 min,取上層清液進樣分析。
糞便樣品:將糞便干燥后研磨粉碎,每0.1 g糞便粉末加入2 mL醇浸提12 h后再超聲15 min,再取出3 500 r/min離心15 min,取上層清液于37 ℃進行氮吹,所得殘渣加入100 μL醇復溶,3 500 r/min離心15 min,取上層清液進樣分析。
1.6 樣品1及M1在大鼠體內的藥代動力學研究
經過“1.3”項研究,在5個種屬的肝微粒體的孵育結果中都找到了樣品1的代謝產物樣品1加氫還原物(M1),經過下文中的二級質譜確證后,推斷出樣品1還原物的結構,然后本課題組合成了該代謝產物,并在大鼠樣品1靜脈注射后進行樣品1及M1的定量研究并計算藥代動力學相關參數。
1.6.1 大鼠頸靜脈插管手術
大鼠用10%水合氯醛(3.5 mL/kg)進行麻醉處理,麻醉后固定于無菌操作臺中,剔除左邊頸部的毛發后,用酒精擦拭干凈皮毛,待大鼠麻醉后,用手術剪剪開皮膚找到頸靜脈,使用1 mm外徑0.5 mm內徑的聚乙烯管插入頸靜脈中縫合固定,后將聚乙烯管從后頸部穿出固定于耳后,縫合后將大鼠靜置24 h,禁食不禁水。
1.6.2 血漿樣品收集
將6只SD大鼠從頸靜脈注射給藥樣品1,給藥劑量為10 mg/kg,分別于給藥前和給藥后0.083、0.25、0.5、1、2、4、8、10、24 h,從頸靜脈插管中取血0.2 mL到EP管中,3 500 r/min離心15 min,分離出上層血漿冷藏于-40 ℃待用。
1.6.3 血漿樣品處理
取大鼠血漿30 μL于1.5 mL的EP管中,加入含20 ng/mL內標SAHA的乙腈120 μL渦旋30 s后13 000 r/min離心15 min,取上層離心液80 μL裝入進樣瓶中進UFLC-MS/MS分析。
1.7 方法學考察
樣品1及M1方法學驗證按照《藥物非臨床藥代動力學研究技術指導原則》進行方法學驗證[12],主要驗證內容有專屬性、線性范圍、精密度與準確度、基質效應、穩定性等。
2 實驗結果
2.1 方法學驗證
2.1.1 專屬性
按照“1.6.3”項下條件,取空白大鼠的血漿30 μL,分別制備空白樣品和含內標和樣品1標準溶液的對照品及含內標和M1標準液的對照品,與實驗血漿樣品進行UFLC-MS/MS分析,進行樣品的專屬性考察。結果顯示:空白樣品無干擾峰,內標SAHA與樣品1及M1分離度好,保留時間分別為1.5、1.99及1.96 min,證明該方法專屬性良好,特異性高(見圖2)。
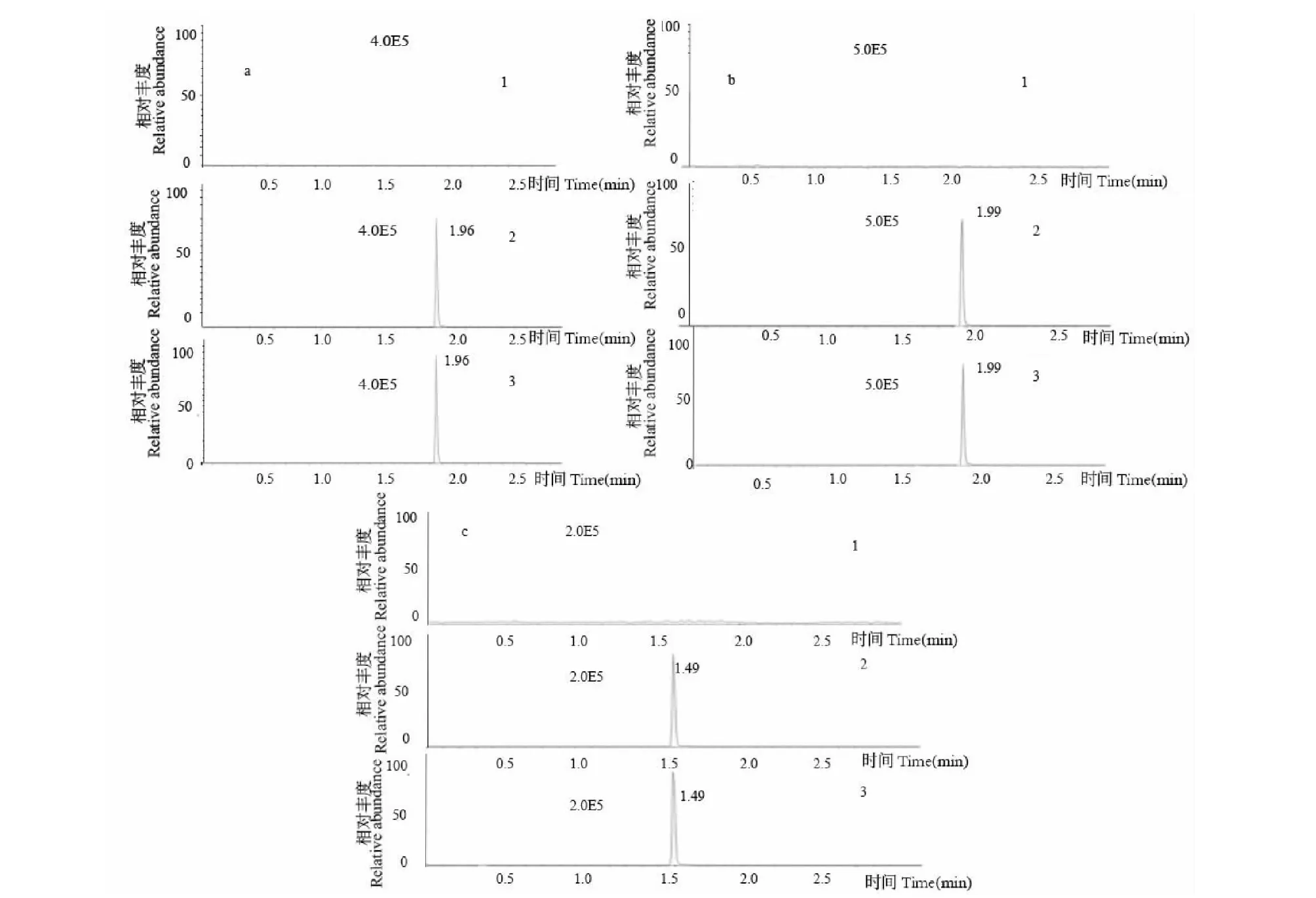
圖2 樣品1、M1分別與內標SAHA的色譜圖Fig.2 Chromatograms of compound 1,M1 and internal standard SAHA注:空白血漿樣品(1);血漿中分別加入樣品1對照品或M1對照品和SAHA(2);給藥后血漿樣品(3)。a:M1;b:樣品1;c:SAHA內標。Note:Blank plasma sample (1);Sample 1 or M1 and SAHA were added to the plasma (2);Plasma sample after administration (3).a:M1;b:Sample 1;c:SAHA internal standard.
2.1.2 線性與范圍
于空白的大鼠血漿中分別加入系列濃度的樣品1、M1標準溶液,終濃度配制成分別為3、10、30、100、300、1 000 ng/mL的標準品溶液,進行UFLC-MS/MS分析,將藥物與內標峰面積比值與藥物終濃度作線性回歸,以加權最小二乘法(權重系數為1/C2),分別考察三條標準曲線,樣品1的結果分別為:y=0.012 04x-0.001 04(R=0.997 01),y=0.0127x+ 0.002 40(R=0.991 30),y=0.012 55x-0.009 86(R=0.994 63),表明樣品1質量濃度在3~1 000 ng/mL范圍內成線性相關,根據信噪比S/N≥10得到最低定量限為3 ng/mL。M1考察三條標準曲線結果分別為:y=0.012 36x-0.006 06(R=0.997 77),y=0.012 21x+0.002 29(R=0.993 60),y=0.011 63x-0.000 04(R=0.996 08),根據信噪比S/N≥10得到最低定量限為3 ng/mL。
2.1.3 精密度與準確度
按照“1.6.3”項下方法,分別配制樣品1及M1質量濃度為9、100、800 ng/mL的低、中、高質量濃度的質控樣品,每個質量濃度質控樣品平行6份,進樣分析,測定樣品1及M1的濃度,考察日內精密度;連續進樣3天,考察日間精密度;同法制備該質量濃度質控樣品,進樣分析,計算各質控樣品測得濃度與理論濃度的回收率,評價該方法的準確度;由表1可知,低、中、高質量濃度質控樣品的日內、日間RSD均小于10%,樣品1的回收率在100.96%~103.21%,M1的回收率在99.20%~105.83%,符合生物樣品定量分析的相關要求[13]。表明該方法精密度、準確度良好,測得結果準確可信。
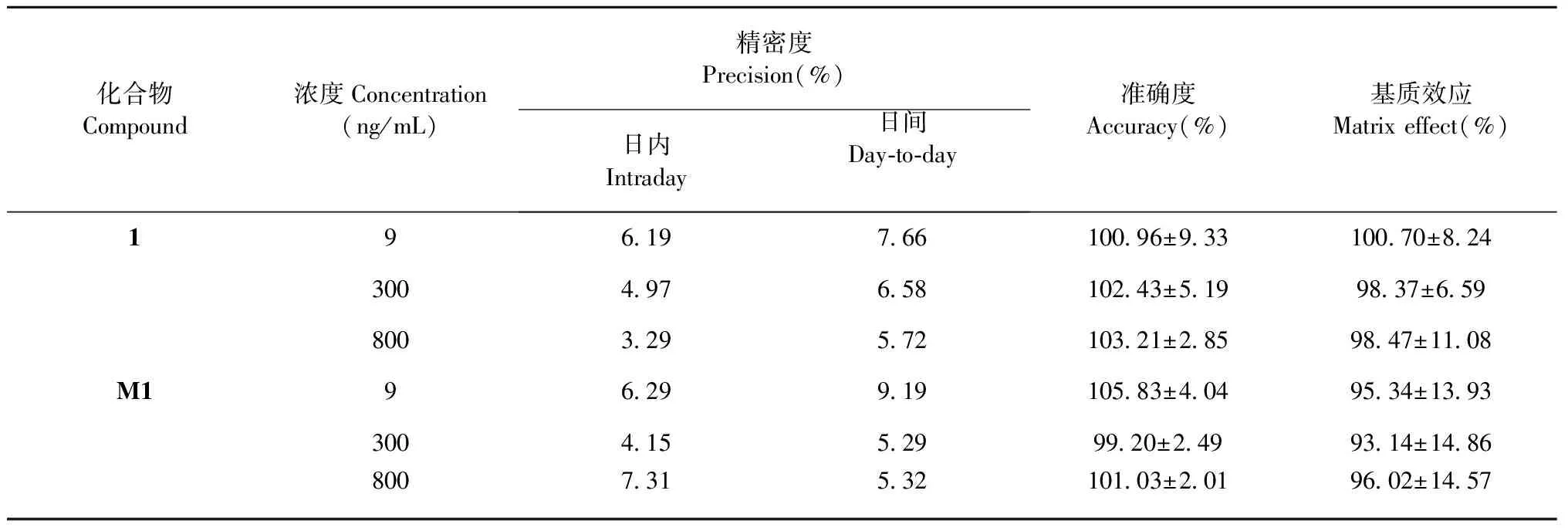
表1 樣品1及M1的精密度、準確度和基質效應測定(n=6)
2.1.4 基質效應
按照“1.6.3”項下方法,用大鼠空白血漿配制質量濃度分別為9、300、800 ng/mL的低、中、高質量濃度的質控樣品,同時用流動相分別配制相同質量濃度的樣品1對照品溶液,進樣分析,分別得峰面積A和B。每組平行5份。基質效應=A/B×100%。由表1結果顯示,樣品1基質效應在98.37%~100.70%,M1基質效應在93.14%~96.02%,偏差小于15%,表明此提取方法無基質效應。
2.1.5 穩定性
按照“1.6.3”項下方法,配制質量濃度分別為9、300、800 ng/mL的低、中、高質量濃度的質控樣品,進樣分析。每組平行5份,考察各質控樣品于自動進樣室內放置24 h的穩定性,-80 ℃放置15天的長期穩定性,以及凍融5次后的凍融穩定性。表2結果顯示在上述各條件下,樣品1及M1樣品穩定性良好。
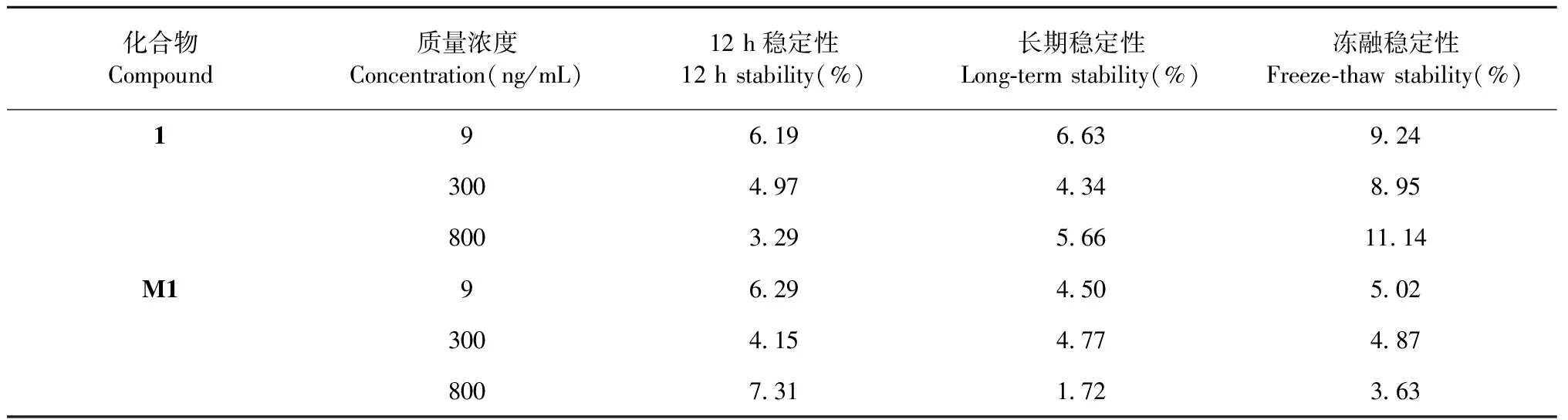
表2 樣品1及M1的穩定性測定(n=5)
2.2 樣品1在不同種屬肝微粒體中的代謝產物
采用一級全掃描正離子方式檢測(MS scan),比較0、60 min兩個時間點的色譜圖差異,初步判斷樣品1的體外代謝產物。結果顯示5個種屬均有2個體外代謝產物:M1(8.36 min),m/z311.127 7([M+H]+);M2(4.06 min),m/z299.128 3([M+H]+)(見圖3)。由于體外代謝產物M1、M2與下文體內代謝產物一致,故在下文體內代謝產物中進行詳細結構分析。
2.3 樣品1在小鼠體內的代謝產物
將處理后的小鼠的血漿、尿液、糞便樣本進UPLC-QE-Orbitrap-MS進行分析,通過比較給藥前后的質譜圖譜,結合各組分的保留時間、精確的一級母離子分子質量、碎片的分子質量等信息,推斷出各個代謝產物的可能結構,預測代謝途徑,在血漿樣本中鑒定出原型藥樣品1及代謝產物M1、M2、M5共4個,在糞便樣本中鑒定出原型藥及代謝產物M1、M2、M3、M4、M5共6個,尿液樣本中鑒定出原型藥及代謝產物M1、M3、M4共4個,各個代謝產物保留時間詳見下圖4中糞便樣本的總離子流圖,各個代謝產物及原藥結構見圖5,各個代謝產物的二級碎片信息見表3。
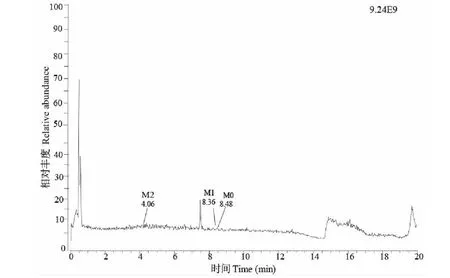
圖3 以人肝微粒體為代表的體外代謝產物總離子流圖Fig.3 Total ion chromatogram of in vitro metabolites represented by human liver microsomes
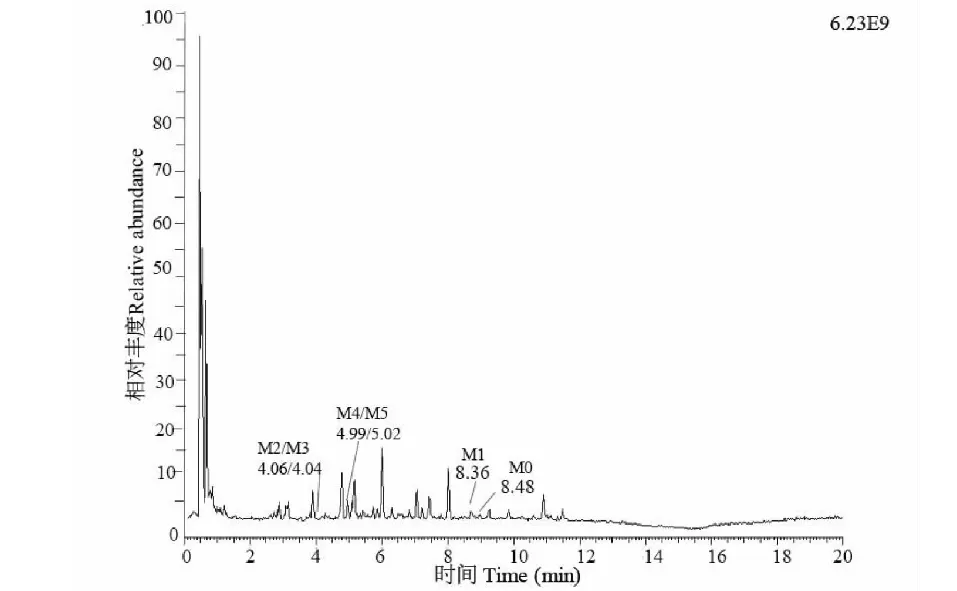
圖4 以糞便為代表的體內代謝產物總離子流圖Fig.4 Total ion current diagram of metabolites in the body represented by feces
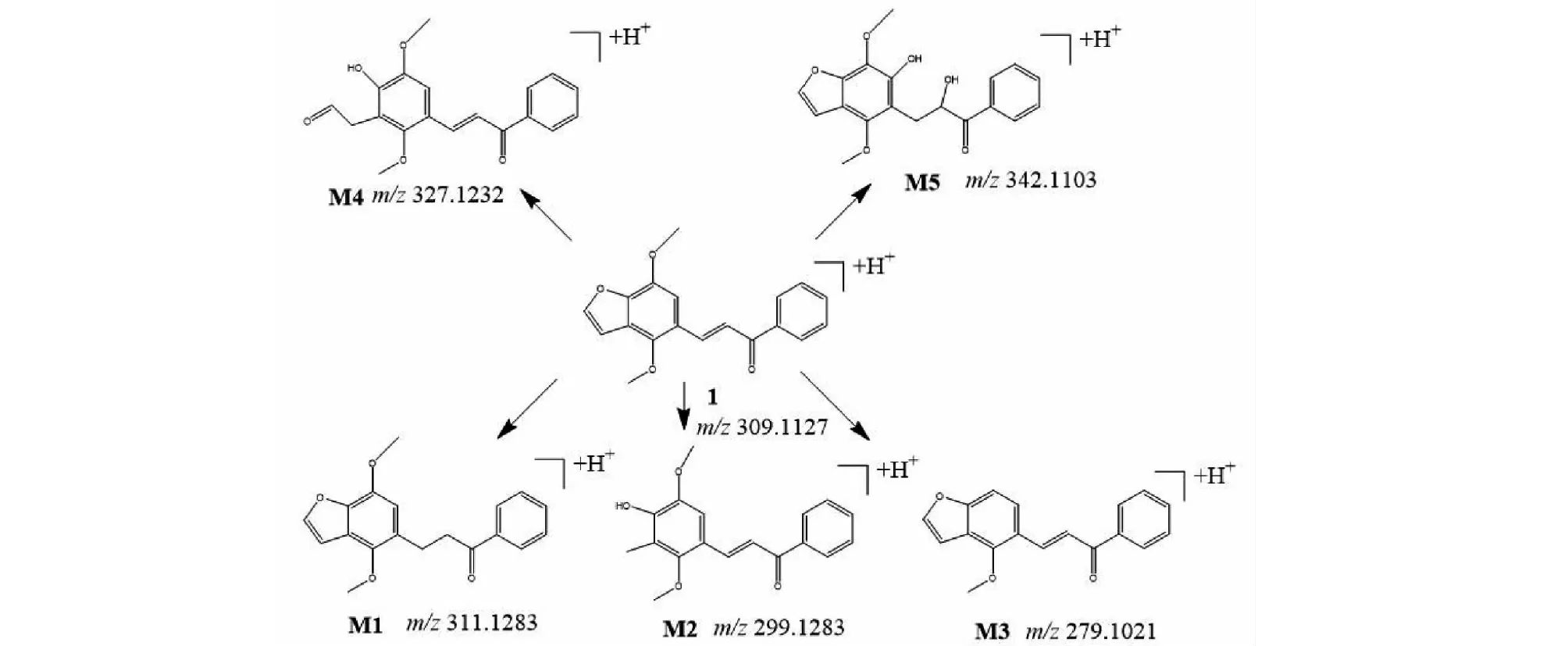
圖5 樣品1及代謝產物產物結構圖Fig.5 Structure of compound 1 and metabolite products

表3 樣品1代謝產物表征
如圖4所示,樣品1(C19H16O4)洗脫時間為8.48 min,準分子離子峰為[M+H]+m/z309.112 7(見圖6a),其二級質譜主要的特征碎片離子有m/z267.101 6 [M-C2HO]+、179.074 0 [M+H-C9H6O]+、131.049 1 [M-C10H9O3]+、105.115 5 [M-C12H11O3]+(見圖7a);可能的裂解途徑見圖8。利用代謝產物與原型藥具有相似的斷裂途徑,m/z131.049 1、178.063 0、105.115 5可作為快速鑒別樣品1體內代謝產物的特征性離子。
M1的準分子離子峰為[M+H]+m/z311.1277(圖6b),推測其分子式為C19H18O4,比化合物1分子質量多2 Da,推斷為化合物1加上兩個H而成。其二級質譜主要的特征碎片離子(見圖9)有m/z269.107 6、191.070 3、105.033 5。其中m/z269.107 6為M1去掉左側呋喃環結構,比化合物1的原藥碎片m/z267.1016多2 Da,其中m/z191.070 3為M1左側的苯環和呋喃環的結構,m/z105.033 5為M1右側的苯環外連接一個羰基的結構,從兩個碎片上看還原所加的兩個H原子未加到兩側的苯環、呋喃環及羰基上,由上推測M1可能為化合物1的中間雙鍵加氫還原代謝產物,后經合成M1后確證。
M2的準分子離子峰為[M+H]+m/z299.127 3(見圖6c),推測其分子式為C18H18O4,比化合物1分子質量少了10 Da,推斷為化合物1結構中呋喃環打開后掉甲基而形成。其二級質譜主要的特征碎片離子(見圖10)有m/z267.101 6、105.033 5。其中m/z267.101 6為M2去掉左側的呋喃環的結構,m/z105.033 5為M2右側的苯環外連接一個羰基的結構,掉的甲基確認在呋喃環上,由上推測M2可能為化合物1的呋喃環打開掉甲基形成。
M3的準分子離子峰為[M+H]+m/z279.101 1(見圖6d),推測其分子式為C18H14O3,比化合物1分子質量少了30 Da,推斷為化合物1結構中去掉甲氧基而形成。其二級質譜主要的特征碎片離子(見圖11)有m/z251.106 7、201.055 1、105.034 0。其中m/z251.106 7碎片可能為M3去掉左側的呋喃環打開后去掉甲基及O原子的結構,m/z201.055 1碎片可能為M3去掉右側苯環的結構,m/z105.033 5為M3右側的苯環外連接一個羰基的結構,由上推測M3可能為化合物1去甲氧基形成。
M4的準分子離子峰為[M+H]+m/z327.122 2(見圖6e),推測其分子式為C19H18O5,比化合物1分子質量多了18 Da,推斷為化合物1結構中加O及2個H原子而形成。其二級質譜主要的特征碎片離子(見圖12)有m/z309.112 2、267.101 3、131.049 4、105.034 0。其中m/z309.112 2碎片可能為M4去掉左側的呋喃環打開后去掉O原子形成,m/z267.101 3碎片可能為M4去掉左側苯環的結構醛基,m/z105.033 5為M4右側的苯環外連接一個羰基的結構,由上推測M4可能為化合物1呋喃環開環形成。
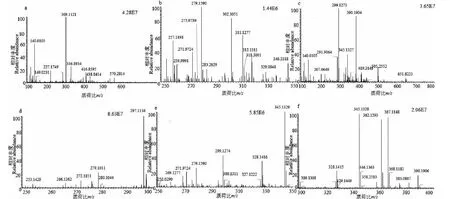
圖6 化合物1及代謝產物的一級色譜圖Fig.6 The MS spectra of 1 and metabolites
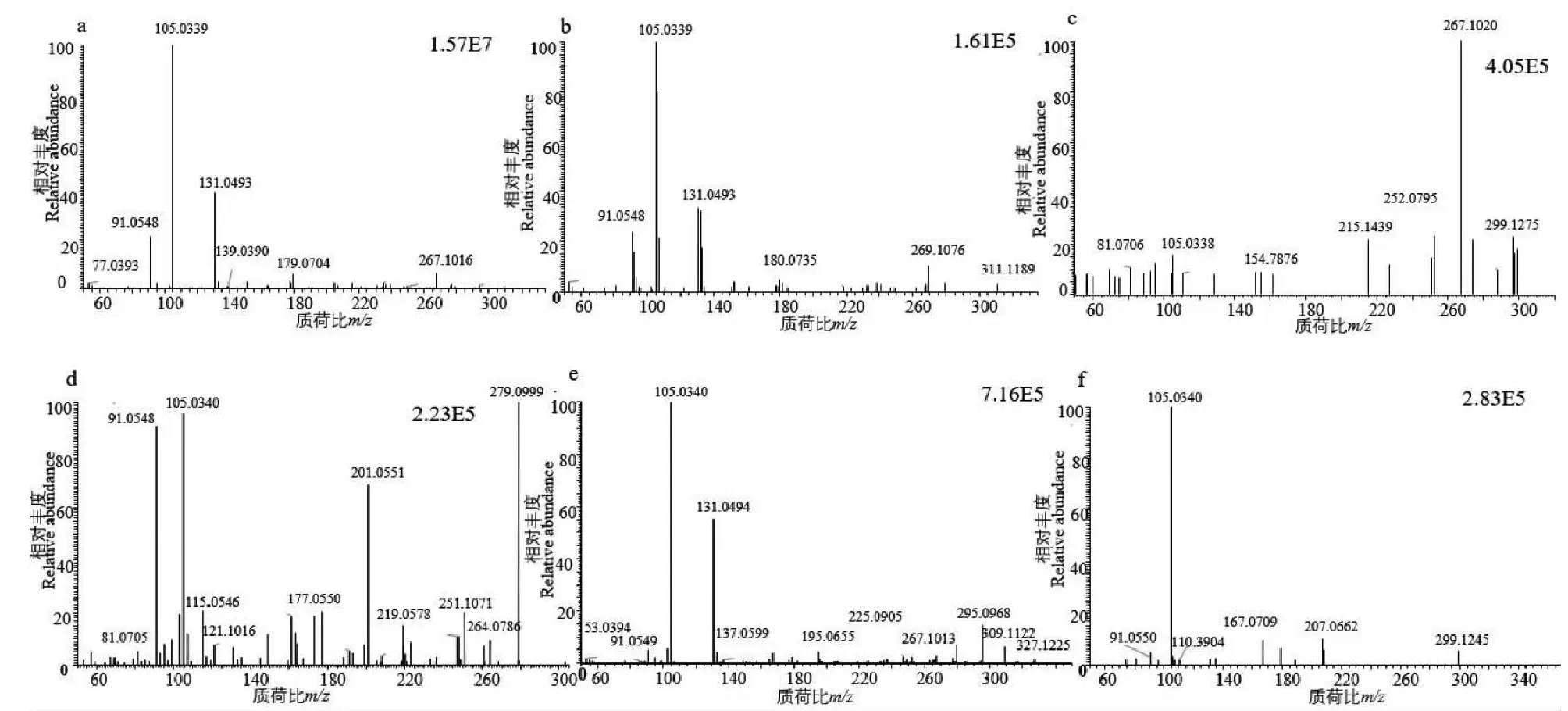
圖7 化合物1及代謝產物的二級色譜圖Fig.7 The MS/MS spectra of 1 and metabolites
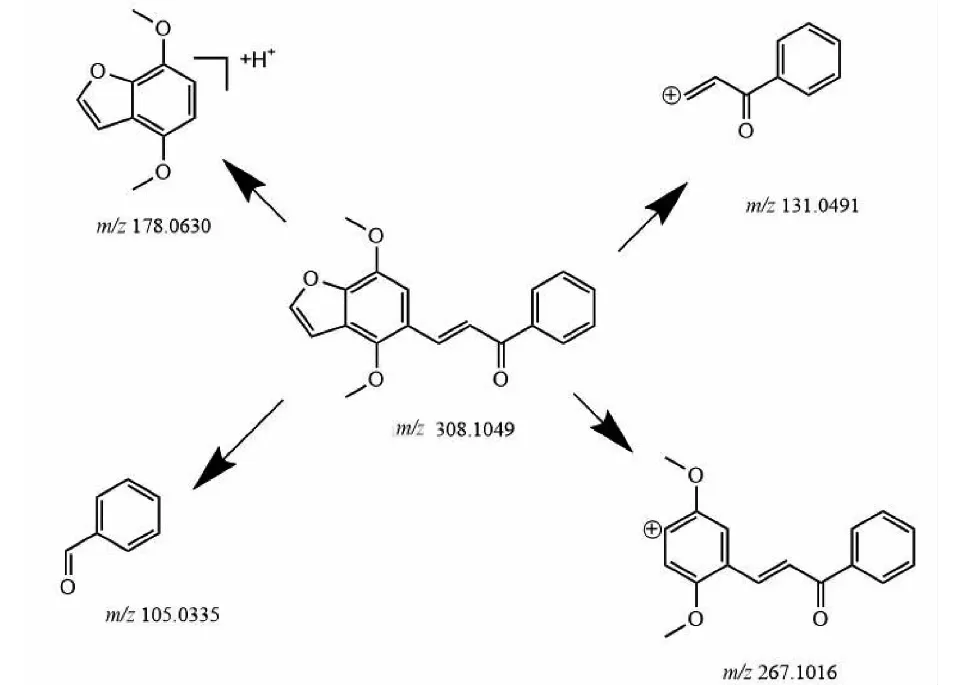
圖8 樣品1可能的裂解機制Fig.8 Possible fragmentation pathways of compound 1
M5的準分子離子峰為[M+H]+m/z343.118 1(圖6f),推測其分子式為C19H18O6,比化合物1分子質量多了34 Da,推斷為M1結構中加2個羥基而形成。其二級質譜主要的特征碎片離子(見圖13)有m/z207.066 2、167.070 3、105.034 0。其中m/z309.112 2碎片可能為M5中間鏈接羥基的碳鏈斷裂形成,m/z167.070 3碎片可能為M5左側苯環上的呋喃環打開形成,m/z105.033 5為M5右側的苯環外連接一個羰基的結構,由上推測M5為M1加兩個羥基形成。
2.4 藥代動力學
按照經過方法學驗證的UFLC-MS/MS方法測定大鼠尾靜脈樣品1給藥后(10 mg/kg)的各個時間點的血藥濃度,采用DAS 2.0進行計算藥代動力學參數,并采用非房室模型對藥物在大鼠體內的動力學過程進行擬合,尾靜脈注射樣品1后,測定大鼠血漿中的樣品1及M1的濃度,藥時曲線見圖14。樣品1的最大血藥濃度Cmax=405.962 μg/L,達峰時間Tmax=0.083 h,半衰期T1/2=3.738 h,藥時曲線下面積AUC0-t=190.635 g/(L·h),清除率Cl=65.578 kg·h/L,表觀分布容積為Vd=299.378 L/kg,M1的最大血藥濃度Cmax=281.291 g/L,達峰時間為Tmax=0.083 h,半衰期T1/2=3.011 h,藥時曲線下面積AUC0-t=561.302 g/(L·h),清除率為Cl=19.179 kg·h/L,表觀分布容積為Vd=79.032 L/kg,結果表明M1在5 min時就達到了最大濃度,樣品1在體內轉換成了代謝產物M1,而后M1濃度一直隨樣品1的減少而減少。
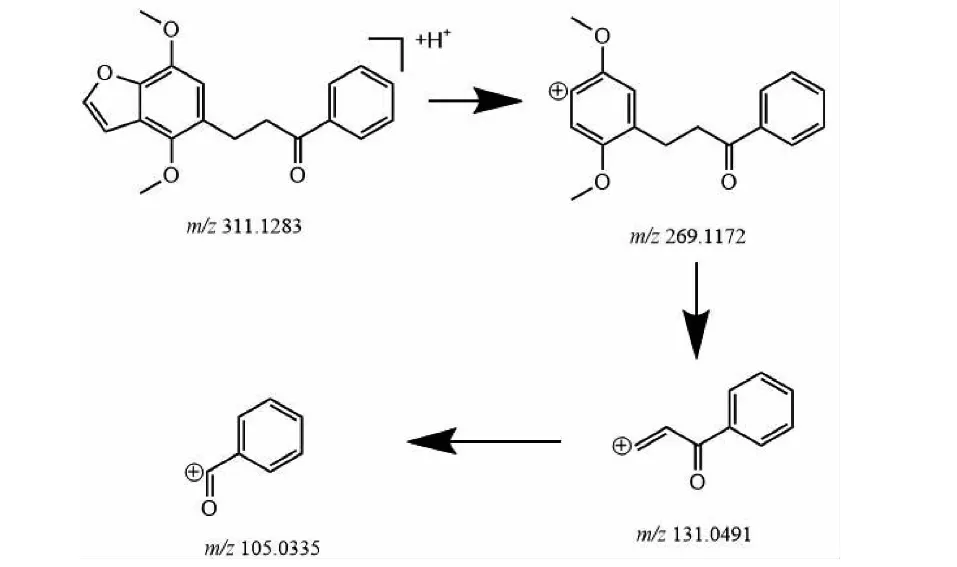
圖9 M1可能的裂解機制Fig.9 Possible fragmentation pathways of M1
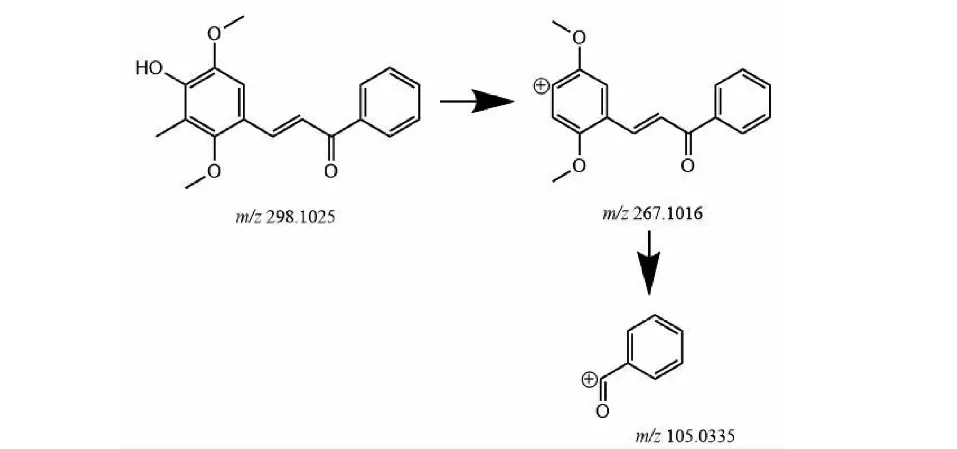
圖10 M2可能的裂解機制Fig.10 Possible fragmentation pathways of M2
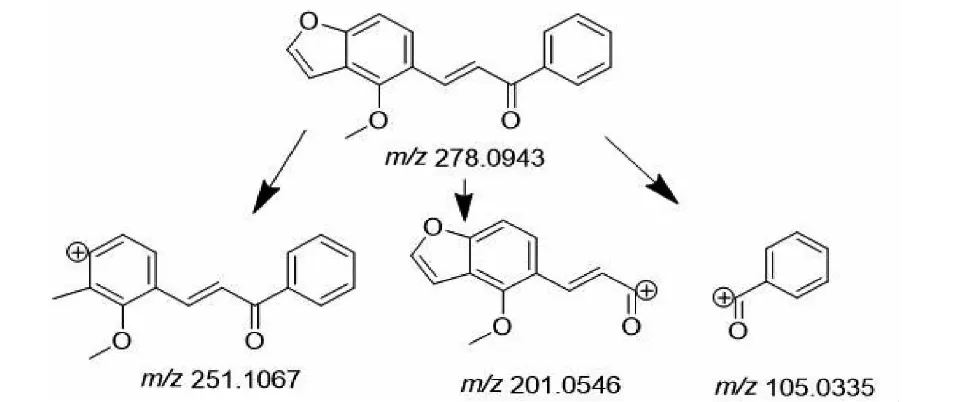
圖11 M3可能的裂解機制Fig.11 Possible fragmentation pathways of M3
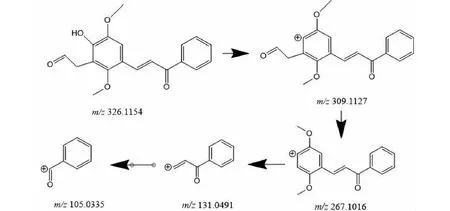
圖12 M4可能的裂解機制Fig.12 Possible fragmentation pathways of M4
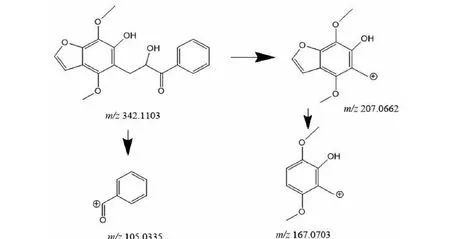
圖13 M5可能的裂解機制Fig.13 Possible fragmentation pathways of M5
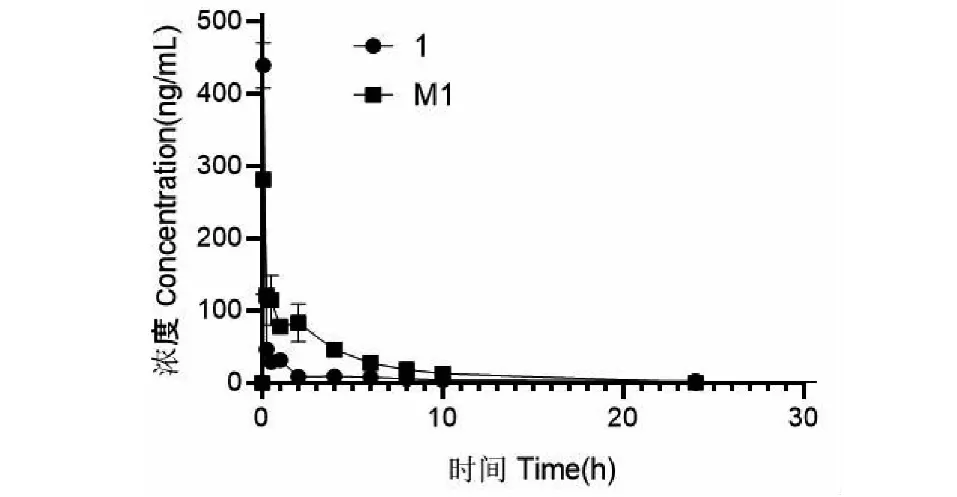
圖14 樣品1及M1在大鼠體內的藥時曲線圖(n=6)Fig.14 Concentration-time profiles of compound 1 and M1 in rats(n=6)
3 討論與結論
絨毛崖豆藤是一味具有良好抗炎療效的中藥之一,樣品1作為其主要的抗炎成分,國內外對其研究較少,尚未見其體內外代謝研究相關報道。本實驗使用UPLC-QE-Orbitrap-MS方法具有較高的靈敏度及專屬性,對于代謝產物的發現及鑒定具有顯著的優勢,同時鑒定出了樣品1在小鼠、大鼠、猴、犬和人5個種屬肝微粒體中的代謝產物為M1、M2,在體內有代謝產物M1、M2、M3、M4、M5,后根據質譜碎片推斷出每個代謝產物的結構,并采用合成的方法制得M1,證明推斷的化合物結構準確可靠。采用UFLC-MS/MS法對體內樣本進行定量分析,通過方法學驗證表明該方法穩定可靠,可以快速定量檢查出樣本中的微量目標化合物,相對于液相色譜法及紫外分析方法具有靈敏度更高等優勢。
化合物的藥代動力學參數是評價化合物的一個重要指標。通過體內外研究其主要代謝產物為M1,后在大鼠體內研究樣品1及M1的代謝速率,并計算其藥代動力學參數,發現樣品1在體內轉換為M1后再進行進一步代謝為M5。本研究對樣品1進行了體內外代謝產物研究,推斷出了5種代謝產物,并在大鼠體內測定了樣品1及主要代謝產物M1的藥代動力學參數,探究了樣品1及M1在大鼠體內血漿中的變化趨勢,結果表明化合物1在體內不穩定,快速代謝為M1,表明起抗炎作用的是其代謝產物而非自身,本研究為其闡明體內抗炎作用物質機制提供理論依據。
猜你喜歡
現代臨床醫學(2022年4期)2022-09-29 07:38:00
昆明醫科大學學報(2021年4期)2021-07-23 01:21:50
天津醫科大學學報(2019年6期)2019-08-13 07:04:34
云南醫藥(2019年3期)2019-07-25 07:25:14
現代檢驗醫學雜志(2016年5期)2016-08-20 03:16:56
海南醫學(2016年8期)2016-06-08 05:43:00
西南軍醫(2016年5期)2016-01-23 02:20:33
川北醫學院學報(2015年5期)2015-12-05 08:22:28
醫學研究雜志(2015年9期)2015-07-01 17:28:15
現代檢驗醫學雜志(2015年1期)2015-02-06 01:59:26
