基于衰減消去蜻蜓算法的小麥粉蛋白質近紅外特征波長優選
2022-08-02 03:11:02吳彩娥熊智新
食品科學 2022年14期
陳 勇,吳彩娥,熊智新*
(南京林業大學輕工與食品學院,江蘇 南京 210037)
小麥粉中蛋白質質量分數約為7%~15%,決定著小麥粉的營養品質和加工品質,所以小麥粉蛋白含量的檢測具有重要意義。小麥蛋白含量測定有化學法和物理法,前者雖然具有較高的準確度,但需要對實驗材料進行繁雜的化學處理,比較耗時費力,同時實驗使用的化學藥品會造成一定的環境污染。與傳統的化學方法相比,近紅外光譜分析是一種綠色、無損快速檢測技術,已經廣泛應用于農業、食品、醫藥、化工等行業。目前,國內外學者利用近紅外光譜法對小麥粉中蛋白質、灰分和水分等含量進行了檢測分析。然而,近紅外光譜有吸收信號弱、譜峰重疊嚴重以及易受外界環境干擾等缺點,并且一條光譜往往包含著數量眾多的波長點,這給建立高質量的預測模型帶來了很大的挑戰。通過合適的波長選擇方法可以在眾多近紅外光譜波長中篩選出具有特征信息的波長,并對數據進行有效地降維,降低建立預測模型的復雜程度,在一定程度上可提高預測模型的準確性。
常見的波長選擇方法有無信息變量消除法、競爭自適應加權采樣法、連續投影算法以及群體智能優化算法。群體智能優化算法具有強大的全局搜索能力,使其在特征變量篩選方面潛力巨大,其中遺傳算法、粒子群算法、灰狼算法等在波長選擇上已有很多成功研究案例。其中,蜻蜓算法(dragonfly algorithm,DA)是Mirjalili等在2016年通過對自然界蜻蜓行為進行觀察、總結和抽象后,提出的一種新智能群體優化算法。之后,分別采用連續DA、二進制DA(binary DA,BDA)及多目標DA,通過對幾類典型函數進行優化驗證了算法的有效性。2019年,Chen Yuanyuan等首次把BDA應用于近紅外特征波長篩選,并比較了單群BDA(single-BDA)、多群BDA(multi-BDA)、基于集成學習BDA(ensemble learning-based BDA,ELB-BDA),表明后兩種算法可以明顯提高波長選擇的穩定性以及分析模型的泛化能力。然而,使用BDA進行波長篩選最終得到的波長位置和波長數量具有強隨機性。盡管使用multi-BDA和ELB-BDA在一定程度上降低了波長數量并進一步篩選出特征波長,提高了算法的穩定性,但是這兩種策略操作復雜,需要多次進行single-BDA運算,導致運行時間遠多于single-BDA,不利于快速建模。并且由于multi-BDA和ELB-BDA的內核還是single-BDA算法,選出的波長分布隨機性仍然較大。為了加快變量選擇速度,Yun Yonghuan等基于達爾文自然進化論中簡單有效的“適者生存”原則,在變量組合集群分析的變量選擇策略中采用指數衰減函數不斷縮小搜尋空間以加快算法速度。借鑒這一思想,在single-BDA算法中引入指數衰減函數,使算法能在迭代過程中由快趨緩地剔除無用變量,有利于保留重要變量,但不利的是由于衰減曲線末端趨向平緩可能造成無效計算,或末端迭代刪除變量數大于1而漏掉某一組或多組變量組合中可能存在的最優變量。為此,本研究嘗試用線性衰減函數代替末期的指數衰減函數,保證算法末期每迭代一次剔除一個變量。Single-BDA算法的改進因此分為兩個階段:第1階段使用指數衰減函數對變量進行快速挑選;第2階段采用線性衰減函數對變量進行精細挑選。本研究把改進算法命名為衰減消去BDA(attenuation elimination-BDA,AEBDA),以期能從小麥粉近紅外光譜中盡快挑選出數量少且穩定的特征波長,并建立精度較高的蛋白質近紅外分析模型。
1 材料與方法
1.1 材料
市購不同品牌以及不同批次的160 個小麥粉的樣品,置于保鮮袋中于室溫(20~23 ℃)保存。
1.2 儀器與設備
Micro NIR Pro 1700便攜式近紅外光譜儀 美國Viavi Solutions公司;D200杜馬斯定氮儀 海能未來科技有限公司。
1.3 方法
1.3.1 小麥粉蛋白質含量的測定
參照GB 5009.5ü2016《食品中蛋白質的測定》中的燃燒法進行測定。
1.3.2 近紅外光譜采集
對小麥粉樣品在室溫(20~23 ℃)環境下不做前處理進行光譜掃描。光譜儀機身采用金屬試管架夾持固定,探頭向下垂直對準深1 cm的圓盤樣品池,樣品池頂部與探頭底部相距1 cm。采集光譜時,小麥粉樣品鋪平樣品池,按120°的角度間隔采集,得到3 條不同檢測點的光譜,取平均作為該樣品的最終采集光譜。儀器的波長范圍為908~1 650 nm,光譜分辨率為6.25 nm,有125 個波長通道,并用與儀器配套的測控軟件MicroNIRTM Pro v2.5采集和儲存信號。
1.3.3 預處理方法
常見的預處理方法有移動平滑法(moving average filter,MAF)、卷積平滑法(Savitzky-Golay filter,SGF)、一階導數(1st D)、標準正態變量變換(standard normal variate transformation,SNV)等。使用這4 種預處理方法的3 種組合(MAF與SNV、SGF與SNV、1st D與SNV,預處理窗口寬度為5)以求達到最好的建模預測效果。
1.3.4 建模與模型評估方法
建模方法采用偏最小二乘回歸(partial least square regression,PLSR)算法,交互驗證采用留一法;single-BDA和AE-BDA算法中采用的適應度函數為PLSR回歸建模中交互驗證標準偏差(root mean square error of cross validation,RMSECV)。
使用RMSECV、預測標準偏差(root mean square errors of prediction,RMSEP)、決定系數()對所建立的校正模型進行評估,計算方法如下:

式(1)~(3)中:y為第個樣品化學值的測定值;y為預測過程中第個樣品的預測值;為所用樣品集樣品數;為化學值測定值的均值。
在建模預測過程中,越接近1,表示模型的回歸或者預測結果越好;如果為負值,表示擬合效果極差。RMSECV與RMSEP的值越小表示所建立的模型的穩定性與精確度越好。
1.3.5 波長變量選擇方法
1.3.5.1 Single-BDA波長選擇
蜻蜓通常通過5 種主要策略改變它們的位置:避撞、結隊、聚集、覓食、躲避。應用到波長選擇方法中,食物即最優解,敵人即極差解,通過不斷改變位置即迭代更新運算直至到達食物的位置即求得最優解。據此建立的位置更新計算方法如表1所示。

表1 蜻蜓5 種主要位置更新策略的數學建模Table 1 Mathematical modeling of five major position shift strategies of dragonfly
采用single-BDA算法進行波長選擇時,位置向量X的每個元素值只能是0或1,因此蜻蜓位置更新并非如表1所示的直接在原有位置X后加上更新速度值Δ,而是只能在0和1之間切換。這就需要對Δ進行連續域到離散域的轉換。最簡單、最有效的方法是采用傳遞函數(式(4)),以Δ作為函數輸入,返回一個[0,1]之間的數值,表示位置變化的概率。

計算所有個體位置的變化概率后,應用式(5)更新蜻蜓在空間中的搜索位置:

式(5)中:為0到1之間的隨機數,負號表示邏輯取反運算。
1.3.5.2 AE-BDA波長選擇
AE-BDA算法是經single-BDA算法結合指數和線性衰減函數改進而來,其在波長篩選過程中主要分為前后兩個部分:快速挑選和精細挑選,兩部分流程如圖1所示。設定最優波長組合比例,AE-BDA算法在迭代運行過程中,統計每次迭代single-BDA計算完畢產生的所有個體中前h個最優RMSECV個體中波長出現的次數。出現的頻率越高,表示波長越重要。然后,按指數或線性衰減函數所計算的當前AE-BDA迭代應保留的變量數,確定需要剔除的無用波長。AE-BDA算法采用均等抽樣法(equal sampling method,ESM),產生算法迭代中single-BDA第一代初始二進制矩陣,保證每個波長具有相同的被選中機會。
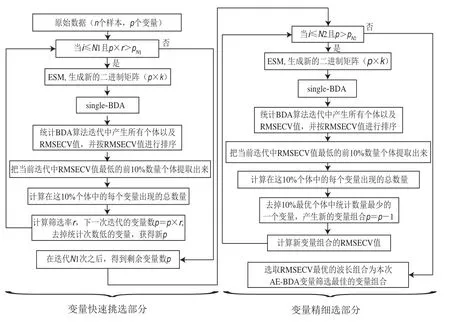
圖1 AE-BDA流程圖Fig.1 Flow chart of AE-BDA
波長快速挑選部分:算法開始,在全波段中無用波長數所占比例高,隨著迭代的進行其所占比例逐步降低,根據指數衰減函數曲線特點,波長剔除速度由快到緩。在這一步中,設置種群數中個體數為,原始變量數為,快速篩選部分篩選變化率,算法運行結束保留的波長個數為p(1為快速挑選部分迭代次數),如果當前迭代變量數hr<p,則快速挑選部分結束;通過不斷的迭代更新,波長數量呈指數級減少,達到所設置保留的波長個數p為止。每次迭代所保留的波長數為:

式(6)中:為當前迭代次數;為波長篩選率;p為第次迭代后所剩的理論波長數。
精細挑選部分:根據線性衰減函數曲線特點,保證在算法末期每代可剔除一個變量。設置最終波長數p(2為精細挑選部分迭代次數),篩選過程中每次迭代并剔除出現次數最少的1 個波長,利用當次迭代得到的波長組合建立PLSR模型,記錄校正集的RMSECV。每次迭代所保留的波長數為:

式(7)中:為當前迭代次數。
當波長數減少到最小設定值p時,迭代終止。選取全部RMSECV記錄中最小的波長組合作為AE-BDA波長優選結果。
圖2中,曲線1為AE-BDA算法波長數量的衰減趨勢圖(以初始變量數=500 個,p=30 個,最終波長數p=10 個為例)。在快速挑選部分,受指數函數性質影響,波長數量經過1次迭代快速降低到p個,然后再由精細挑選進一步壓縮波長數量至最小波長數p個;曲線2為指數衰減函數變量衰減曲線,曲線1相比較于曲線2在算法末端(p至p階段)逐個波長剔除,不會導致某一波長點數被跳過。圖2的柱狀圖為每一次迭代剔除的無用變量數量,在快速挑選部分,隨著迭代次數的增加每次迭代剔除的無用變量數也呈指數減少;在精細挑選部分,每一代消去1 個無用變量,保證無用變量被精細均勻剔除。
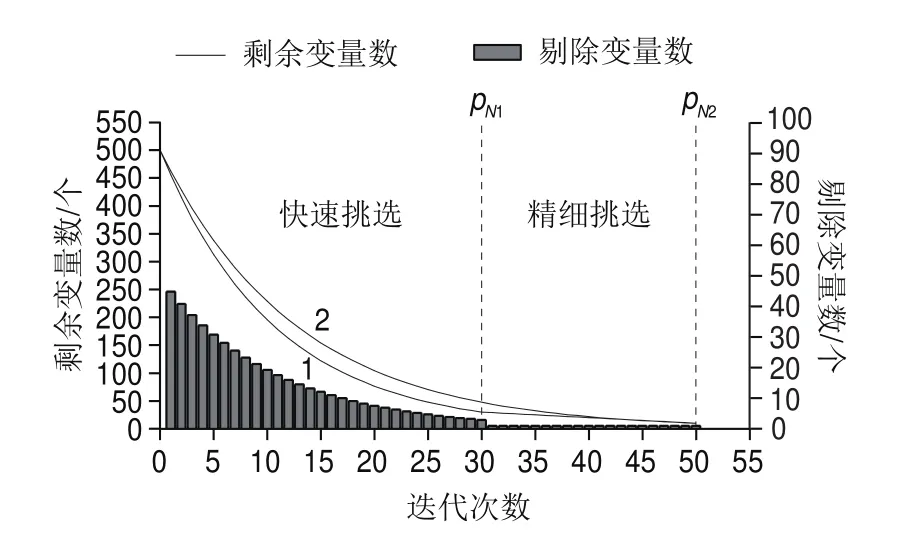
圖2 AE-BDA算法波長數衰減圖Fig.2 Decreasing number of variables in AE-BDA
1.4 數據處理
使用MATLAB 2016a軟件以及實驗室自主開發的NIRSA 5.9.4化學計量學軟件,采用主成分分析和馬氏距離相結合的方法檢測異常樣本,主成分分析結合Kennard-Stone的方法對校正集和預測集進行劃分。
2 結果與分析
2.1 小麥粉近紅外光譜圖
圖3為小麥粉近紅外光譜部分原始光譜,共有160 條小麥粉近紅外光譜,每條光譜含125 個波長通道。
圖3 小麥粉近紅外光譜部分原始光譜Fig.3 Selected original near-infrared spectra of wheat flour
2.2 小麥粉蛋白質含量
由表2可知,所選樣本的蛋白質含量基本覆蓋小麥粉中蛋白質質量分數(7%~15%)且分布較為均勻,表明實驗所選樣本具有代表性。

表2 小麥粉蛋白質含量統計表Table 2 Statistics of protein content of wheat flour
2.3 樣本異常值剔除與樣本集劃分
樣品、采集環境和儀器在一定程度上會產生異常樣本數據,嚴重影響所建模型的穩定性與預測能力,所以在建模前必須將異常樣本數據從集合中剔除。3 種預處理組合各自檢測出的異常樣本集的并集(共8 個樣本)作為總異常樣本集予以剔除。最后,劃分剩余的152 個樣本為校正集和預測集;共得到98 個校正集樣本,54 個預測集樣本。如表3所示,校正集與預測集的樣本分布較寬,具有良好的代表性。
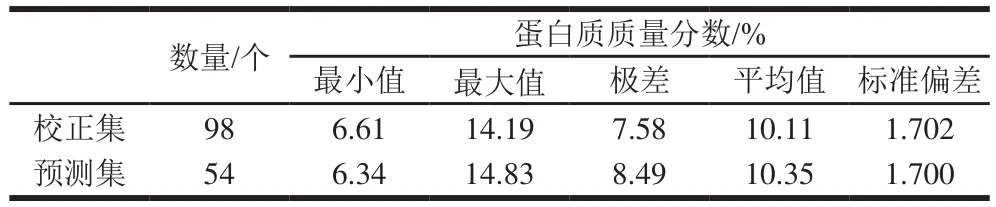
表3 校正集與預測集小麥粉蛋白質含量統計表Table 3 Statistics of protein content of wheat flour in calibration and prediction sets
2.4 預處理方法對全譜段PLSR建模及預測的影響
由表4可知,MAF與SNV預處理組合的PLSR模型除
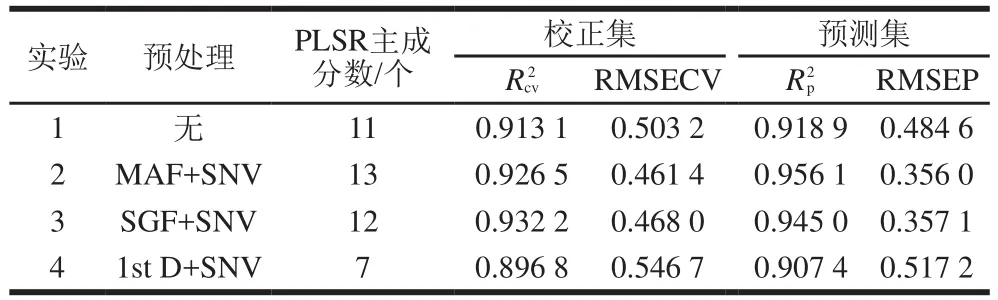
表4 不同預處理方法全譜PLSR模型性能Table 4 Performance of full-band PLSR models developed using different preprocessing methods
值略低于SGF與SNV預處理組合的PLSR模型外,各指標均優于其他建模方法。這說明采用MAF和SNV組合的預處理方法建立PLSR全譜校正模型可以提高小麥粉近紅外全譜段模型穩定性和預測精度。因此,后續波長選擇算法研究中光譜都采用MAF與SNV組合進行預處理。
2.5 2 種算法對波長選擇建模及預測的影響
分別用single-BDA和AE-BDA 2 種方法進行波長篩選后建模,16 次建模及預測實驗結果的比較列于表5。實驗S1~S8波長選擇方法為single-BDA,實驗A1~A8波長選擇方法為AE-BDA。其中,AE-BDA算法設置種群數為200 個,衰減率為0.9,p設置為20 個,p設置為10 個,每次迭代中內核single-BDA迭代次數為10 次。經過程序運行后AE-BDA共迭代28 次(快速挑選迭代18 次,精細挑選迭代10 次),其內核single-BDA共迭代280 次。為了便于改進前后方法對比,single-BDA設置種群數量為200 個,迭代次數為280 次,篩選率為20%。
由表4和表5可知,無論是single-BDA還是AE-BDA算法,挑選后的波長進行建模及預測效果均明顯優于全譜段建模。使用single-BDA進行波長選擇時,實驗S1~S8所挑選出的平均波長數為28.38 個,占原始波長數(125 個)的22.7%;而使用AE-BDA方法進行波長選擇時,實驗A1~A8所挑選出平均波長數為15.8 個,為原始波長數的12.6%,相較于single-BDA方法篩選出建模波長數量更少。同時可以看到,AE-BDA方法挑選出的波長建模和預測評價指標相應都較接近,且其預測效果總體上好于single-BDA方法。盡管single-BDA方法建立的模型評價指標及RMSECV總體上比AE-BDA好,但預測效果卻相對略差,這說明single-BDA方法選擇的波長過多,建立的模型可能存在過擬合現象,導致預測結果變差。
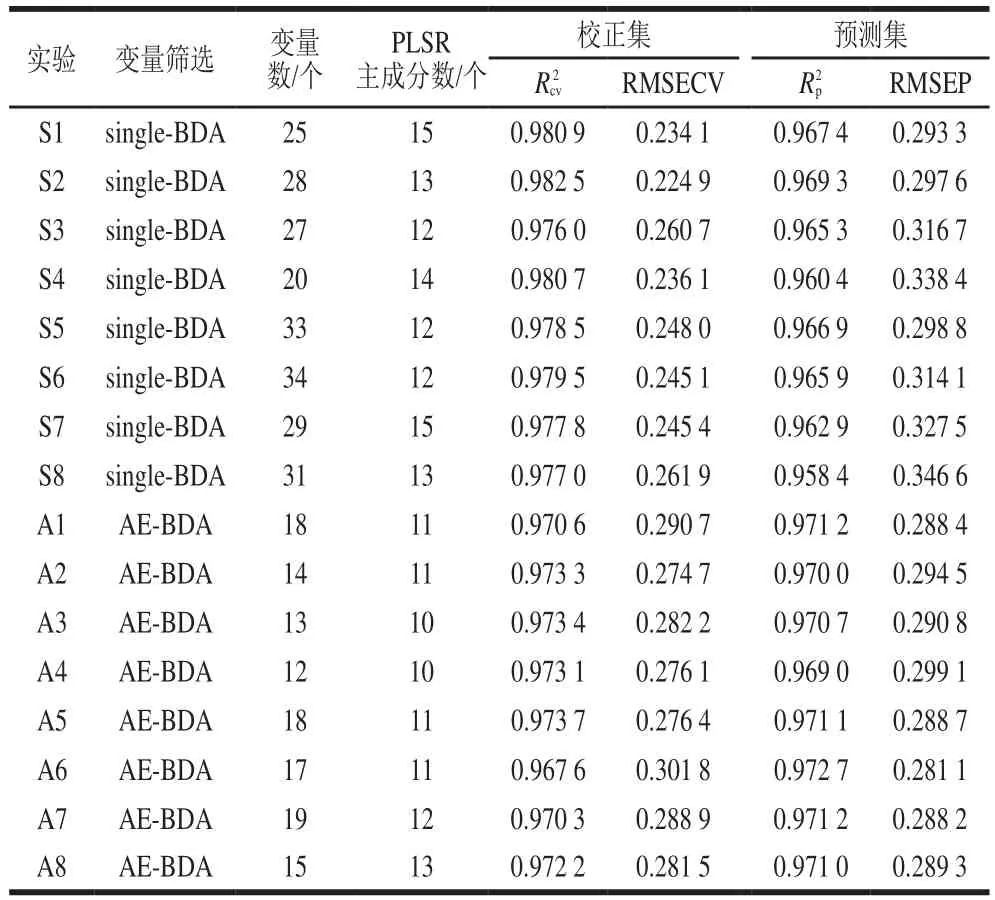
表5 Single-BDA與AE-BDA建模及預測性能比較Table 5 Comparison of modeling and prediction performance between single-BDA and AE-BDA
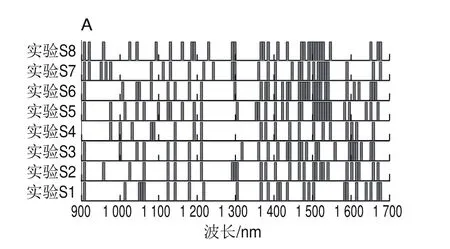
由圖4和圖5可知,盡管兩種方法都是采用隨機算法更新決策變量,但AE-BDA與single-BDA相比所挑選的波長點少,分布范圍更加集中,且AE-BDA中有3 個特征波長(1 385.07、1 508.95 nm和1 589.48 nm)在8 次實驗中均被挑選出來,而single-BDA方法挑選出頻次最多的2 個波長(908.1、1 502.76 nm)只有7 次。這說明AE-BDA挑選波長的隨機性要弱于single-BDA方法,結果更加穩定。

圖4 Single-BDA(A)和AE-BDA(B)實驗入選波長位置Fig.4 Selected wavelength locations in single-BDA (A) and AE-BDA (B) experiments
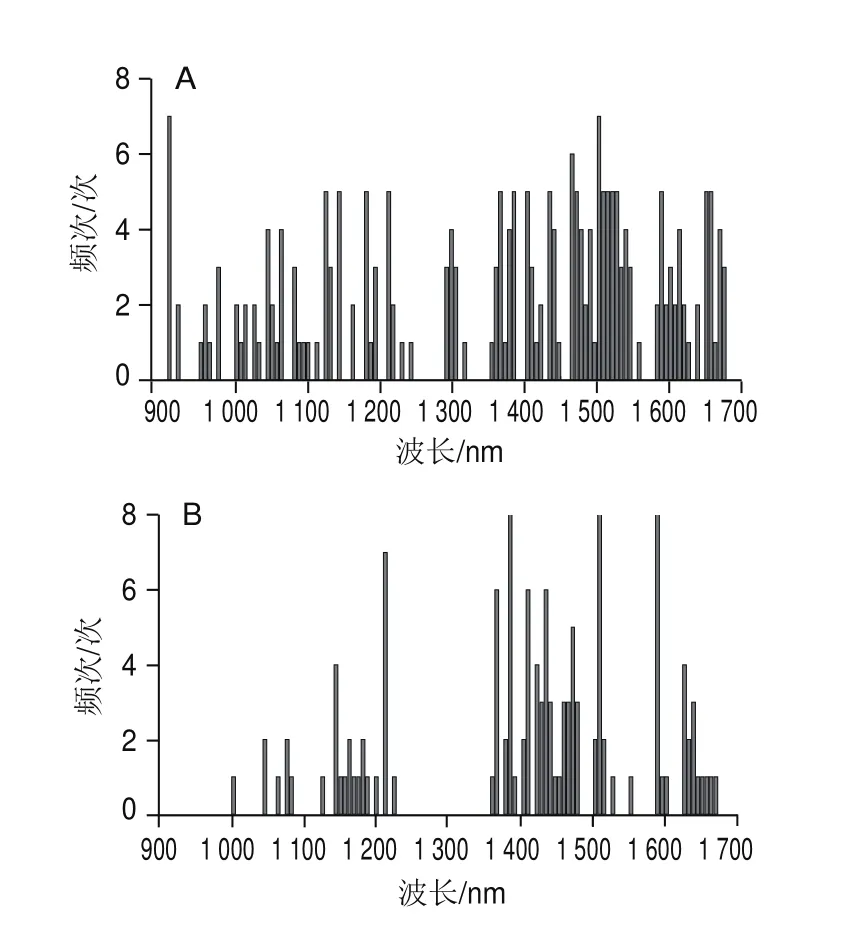
圖5 Single-BDA(A)和AE-BDA(B)實驗中波長出現頻次圖Fig.5 Frequency of appearance of wavelengths in single-BDA (A) and AE-BDA (B) experiments
AE-BDA相較于single-BDA方法計算效率更高。由圖6可知,種群數相同(200 個)的情況下,隨著迭代次數的增加,AE-BDA參與迭代計算的總波長數呈指數下降,意味計算量也呈指數級降低,計算過程逐步加速;而single-BDA算法在迭代過程中,進入每一次迭代的總波長數是不變的,計算量不變,運算速度保持相對恒定,運算時間較長。
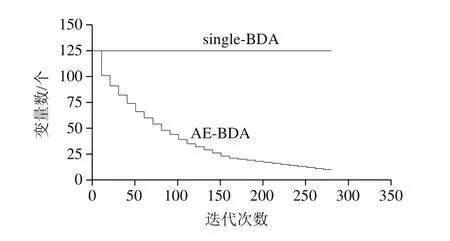
圖6 Single-BDA和AE-BDA在迭代過程中初始波長數變化曲線Fig.6 Change in initial number of wavelength in single-BDA and AE-BDA as a function of number of iterations
雖然近紅外光譜帶重疊嚴重且很難清晰地指出各譜峰對應的具體基團信息,但根據近紅外主要譜帶歸屬仍可對所選波長的合理性做出必要分析。本研究僅討論8 次AE-BDA實驗中每次都被挑選出的3 個波長點。蛋白質分子中含有氨基,NüH鍵伸縮振動的一級倍頻吸收帶位于1 510 nm波長附近,1 508.95nm波長處為-氨基酸中的氨基的吸收峰;甲基和亞甲基中CüH鍵的伸縮振動的一級倍頻和變形振動的基頻吸收帶位于1 360~1 395nm波長處,1 385.07 nm波長處為CüH吸收峰;波長1 589.48 nm處位于OüH鍵的一級倍頻吸收譜區,由于模型中其標準回歸系數為負且較大,可以判斷為主要由非蛋白質分子中OüH鍵吸收引起(波長點位置見圖7)。這可以作為對小麥粉中淀粉、脂肪等強背景組分的修正。通過進一步對照分析可以看出,所選其他波長點大部分也位于以上譜帶區域內或者附近。
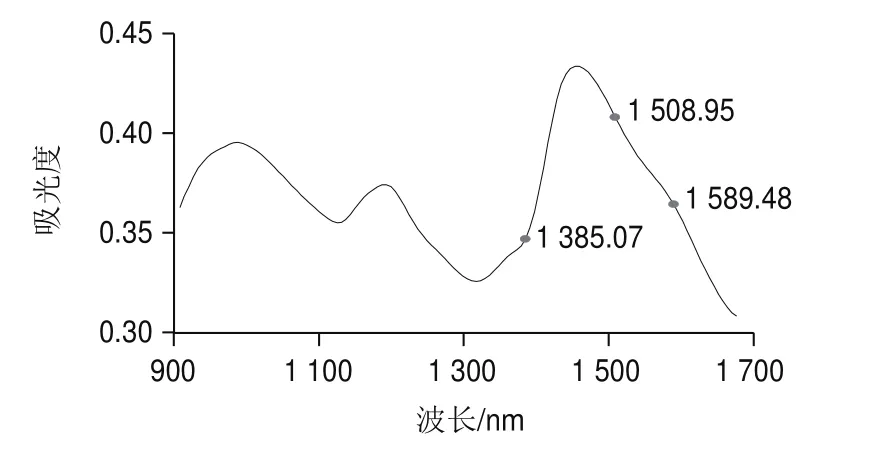
圖7 出現8 次的波長點在全譜段的位置Fig.7 Locations of wavelengths that appeared eight times in the full-band spectrum
3 結 論
針對常規的single-BDA在近紅外光譜波長篩選過程中出現的問題,引入指數衰減函數和線性衰減函數對其進行改進,形成AE-BDA,并結合PLSR建立了小麥粉蛋白質近紅外分析模型。通過對AE-BDA所挑選出的波長進行特征分析表明,這些波長較好地分布于蛋白質分子的主要官能團吸收區域。蛋白質分子中重要的NüH鍵的吸收波長(1 508.95 nm)每次都能被挑選出來;其他反映蛋白質分子的甲基、亞甲基以及次甲基的CüH鍵吸收波長也因出現頻次高而作為挑選出的特征波長用于建立小麥粉蛋白質近紅外分析模型,提高了模型的可解釋性。
改進后的AE-BDA運算速度快,所選波長穩定,且一定程度上克服了過擬合,所建模型精度高。這為小麥粉蛋白質近紅外光譜提供了一種可靠、有效的波長挑選方法,對于其他分析對象的近紅外光譜的變量選擇也有重要的借鑒意義。在后續研究中可以優化算法中各因子的取值范圍,探討其對AE-BDA的影響,進一步提高算法的運行效率和自動化水平。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
兒童故事畫報(2019年5期)2019-05-26 14:26:14
光學精密工程(2016年6期)2016-11-07 09:07:19
Coco薇(2016年2期)2016-03-22 02:42:52
核科學與工程(2015年4期)2015-09-26 11:59:03
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
