[Ir(ppy)2(py-x)]+配合物的密度泛函理論研究
2022-08-10 03:13:32聶建航金雨廷張建坡
吉林化工學院學報 2022年3期
聶建航,王 哲,金雨廷,張建坡,金 麗
(吉林化工學院 化學與制藥工程學院,吉林 吉林 132022)
在數十年的研究中,過渡金屬配合物發光材料由于在OLED技術中的廣泛應用[1-3],越來越多的OLED產品相繼問世,特別是能提升發光效率、壽命以及拓展發光區域的新材料開發具有重要意義.而在一系列過渡金屬中,磷光陽離子銥(Ш)配合物因其顯示出覆蓋整個光譜的可調激發和發射波長以及獨特的光物理特征引起了社會的廣泛關注,使其具有重要的科研價值和應用前景[4-5].
有研究表明,在激發態Ir與雙(吡唑-1-基)甲烷的結合比主要的苯基吡啶配體弱得多,雙(吡唑-1-基)甲烷的存在確保電子進一步定位在主要的苯基吡啶配體上,這導致三線態發射相對于以前使用芳香輔助配體的復合物顯著藍移[6].為了進一步探究含有雙(吡唑-1-基)甲烷配體的Ir配合物的發光規律,選取一類[Ir(ppy)2(py-x)]+配合物采用量子計算方法進行理論探究,考察雙(吡唑-1-基)甲烷配體上取代基的差異對分子結構和發光性質的影響.
1 計算方法
在曙光A620-G30服務器上使用Gaussian09程序包完成全部計算.使用密度泛函[7](Density functional theory)水平下的B3LYP[8]泛函和單激發組態相互作用(CIS)[9]方法分別優化了配合物基態和激發態幾何結構,依據Franck-Condon垂直躍遷原理應用含時密度泛函[7]方法結合PCM[10](Polarized continuum modle)溶劑化模型計算了[Ir(ppy)2(py-x)+]配合物1-3在CH2Cl2溶液中的吸收和發射情況.對Ir原子使用只考慮價電子的贗勢LANL2DZ[11]基組計算,對C、N、H原子使用6-31 G基組計算.該方法對于此類配合物的計算已被證實是可靠的.
2 結果和討論
2.1 基態和激發態結構
圖1為3個分子的基態結構圖,相對應的基態和激發態幾何參數還有實驗值都在表1體現.從表1中可以看出,3個配合物都具有1A基態和3A激發態.配合物1-3的基態Ir-N(1)、Ir-N(4)、Ir-N(6)鍵長與實驗值之差都在0.11?范圍內,鍵角N(1)-Ir-N(4)與實驗值相差2.5 °,計算值和實驗值的偏差處于合理范圍內.激發態時,選擇與Ir相連的4個主要鍵長都有所延長,表明激發態時電子躍遷由金屬向配體躍遷,削弱了金屬和配體之間的相互作用,且激發態時鍵角和二面角變化都不大.由表1中列出的3個分子主體結構數據可得出,吡咯、吡唑配體的引入并未對配合物主體結構造成較大影響.
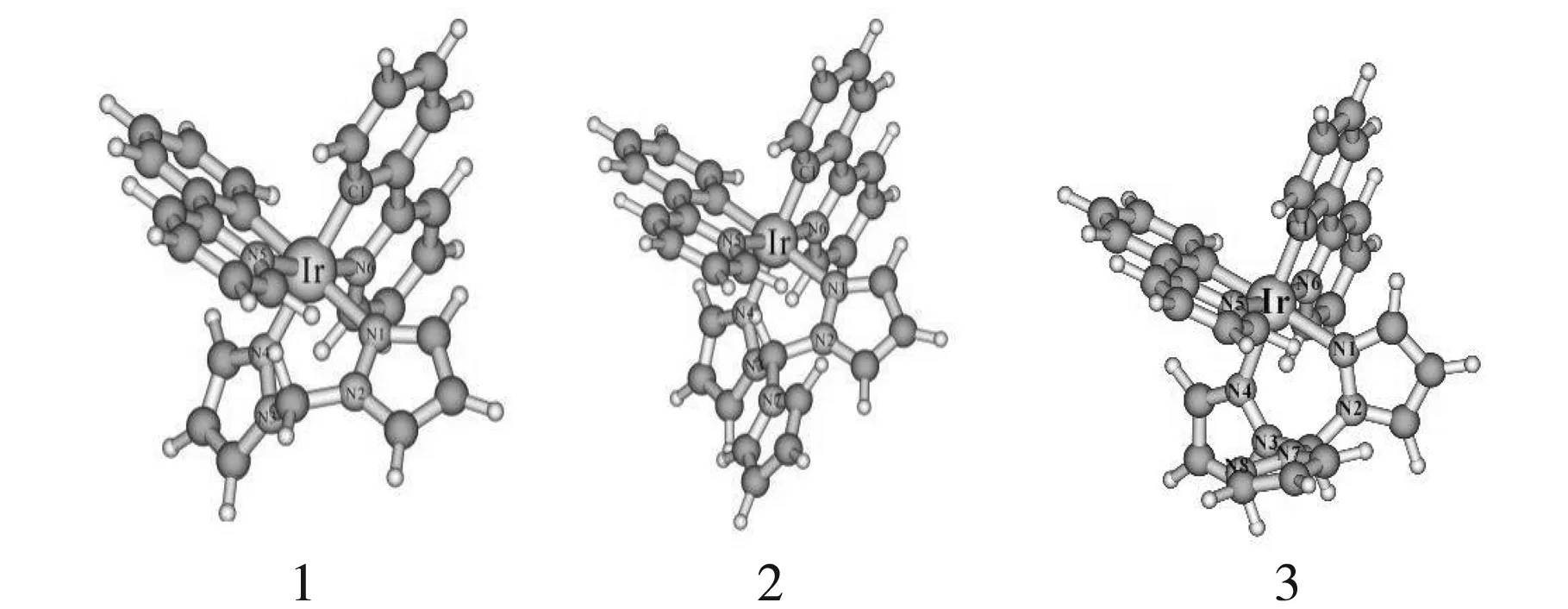
圖1 配合物1-3的優化結構簡圖

表1 分別用B3LYP和CIS方法計算得到配合物1-3基態以及激發態的主要幾何參數
2.2 配合物的吸收光譜
在CH2Cl2溶劑中,分別記錄了分子1、2、3以TD-DFT(含時密度泛函)方法得到的Gaussian型基態吸收光譜如圖2所示.

Wavelength/nm圖2 配合物1-3在CH2Cl2溶液中模擬的Gaussian型吸收曲線
在表2中給出了選擇的主要吸收和發射數據,包括躍遷軌道、波長、能量、振蕩強度和所屬性質.在表3中陳列出其提及的分子軌道相關成分和能量.

表2 配合物1-3在CH2Cl2溶劑中的吸收和發射光譜數據

表3 配合物1-3在B3LYP泛函水平下的基態分子軌道成分
從表2和圖2中能夠發現,3個分子都有兩個高強度吸收帶和1個強度較小最低能吸收帶.配合物1-3的最低能吸收分別為399.07(3.11),401.68(3.09)和396.15(3.13) nm,配合物1的399.07 nm的吸收來自分子軌道128→129的激發,軌道128為最高占據軌道,主要由48.1%的d(Ir)和48.2%的π(ppy)構成,而129為最低空軌道,主要由90.3%的π*(ppy)配體占據.因此,該躍遷歸屬于d(Ir)+π(ppy)→π*(ppy)的金屬到配體和ppy配體內部的電荷轉移(MLCT/ILCT)躍遷.配合物2的401.68 nm的吸收也是來自HOMO到LUMO的躍遷,并且與配合物1有相似的躍遷性質,但配合物3略有不同,它的396.15 nm的吸收來自分子軌道145(HOMO)到146(LUMO)的激發,HOMO軌道主要由45.1%的d(Ir)和51.4%的π(ppy)構成,LUMO軌道由40.3%的π*(ppy)和54.1%的π*(py-pz)構成,因此該躍遷僅屬于d(Ir)+π(ppy)→π*(ppy)+π*(py-pz)的金屬到配體py-pz的躍遷,ppy配體成分在躍遷前后基本不變.通過分析3個配合物的最低能吸收波長可以發現,吡咯的引入,使配合物2的最低能吸收波長發生較小的紅移(2.61 nm),吡唑的引入,使配合物3的最低能吸收波長發生較小的藍移(2.92 nm).
相對于最低能吸收,其他兩個吸收帶明顯增強.觀察圖2,第2吸收帶分布在301 nm到327 nm之間的寬闊區域,第3吸收帶為高能吸收帶,振子強度最大,集中分布在260 nm到272 nm之間,波長相差較小.不同配體的引入對吸收帶的位置產生了一定影響.根據上文表2、3可知,吸收帶2分別由HOMO-2→LUMO、HOMO-4→LUMO+1、HOMO-3→LUMO+1貢獻,且躍遷屬性都歸屬于MLCT/ILCT躍遷.吸收帶3分別由HOMO-3→LUMO+3、HOMO→LUMO+4、HOMO+4→LUMO+4激發捐獻.以分子1為例,HOMO-3由41.8%金屬Ir和49.7%的ppy配體π成鍵軌道占據,LUMO+3是由75.2%的py-H配體和20.5%的ppy配體構成的π反鍵軌道占據,經分析得出此躍遷被指認為MLCT/LLCT混合躍遷.同理分析得出配合物2、3在該位置擁有與配合物1相似的躍遷屬性.
2.3 配合物的磷光發射光譜
配合物1-3在CH2Cl2溶液中的磷光光譜數據如表2所示,3個分子的磷光發射分別位于513.00 nm(1)、513.78 nm(2)、510.38 nm(3).他們都起源于HOMO→LUMO的激發,分子1、2的HOMO軌道主要占據在金屬Ir和ppy配體上,而LUMO軌道只由ppy配體捐獻,因此最低能吸收具有相似的躍遷屬性,但配合物3略有不同,其LUMO由40.3%的py-pz配體和54.1%的ppy配體構成.比較配合物1-3的最低能吸收和發射(HOMO→LUMO),配合物1、2具有相似的軌道成分和躍遷性質(MLCT/ILCT),配合物3則因吡唑的引入略有不同(MLCT).能量差分別為0.69、0.68和0.70 eV,其斯托克斯頻移非常接近,表明取代基的引入對此類配合物的影響較小.
3 結 論
本文從理論上對配合物[Ir(ppy)2(py-H)]+(1),[Ir(ppy)2(py-pr)]+(2),[Ir(ppy)2(py-pz)]+(3) 的基態以及激發態結構、軌道占據、光譜性質和躍遷屬性進行了詳細的研究.結果表明:HOMO軌道占據成分不受引入吡咯、吡唑配體的干擾,但LUMO軌道成分在引入吡唑配體后發生很大變化,分子1的LUMO主要為90.3%的π*(ppy),分子2的LUMO主要為89.3%的π*(ppy),而分子3的LUMO主要為54.1%的π*(ppy)和40.3%的π*(py-pz),從而使躍遷性質發生根本性的改變.吡咯和吡唑的引入都使HOMO和LUMO的軌道能和躍遷波長發生明顯改變,吡咯的引入使分子2的吸收和發射紅移,吡唑的引入使分子3的吸收和發射藍移,但總體移動趨勢都比較小,這主要是由于雙(吡唑-1-基)甲烷配體本身為非共軛配體,在甲基部分引入取代基團依然不能產生共軛效應,因此對配合物的吸收和發射影響都不大,要想改變配合物的發光顏色需要圍繞ppy配體來進行分子設計.
