基于LASSO回歸和多層感知的癌組織RNA-Seq數(shù)據(jù)分類算法研究
2022-08-31 23:35:23顏瀅李文敬李松釗
電腦知識與技術 2022年19期
關鍵詞:特征提取
顏瀅 李文敬 李松釗


摘要:目的:為了解決癌癥基因RNA-Seq(RNA-Sequencing,轉錄組測序技術)技術每次測序過程產(chǎn)生海量高分辨率、高維、高冗余的數(shù)據(jù),給基因表達數(shù)據(jù)分類帶來困難的問題。方法:提出了一種基于LASSO(Least Absolute Shrinkage and Selection Operator,LASSO)回歸和多層感知的癌組織RNA-Seq數(shù)據(jù)分類算法。首先,從TCGA數(shù)據(jù)庫獲取十個疾病的基因數(shù)據(jù)集并對原始RNA-Seq的基因表達譜基因數(shù)據(jù)進行數(shù)據(jù)清洗和標準化處理,去除重復的基因,選取表達量最大的基因并將數(shù)據(jù)做標準化處理。其次,采用LASSO回歸的方法對處理后的數(shù)據(jù)進行降維和特征提取,獲得與疾病標簽最相關的特征基因集。最后,運用多層感知器神經(jīng)網(wǎng)絡(Multilayer Perceptron,MLP)模型對特征基因進行學習和訓練,實現(xiàn)有效地識別和分類。實驗結果:實驗表明,該算法在10種癌細胞基因測試數(shù)據(jù)集中分類總準確率達到99.8%,高于LASSO-CNN分類模型的總準確率98.9%和LASSO-BP神經(jīng)網(wǎng)絡分類模型的總準確率99.4%。結論:該算法克服了轉錄組測序數(shù)據(jù)量大、特征多、數(shù)據(jù)差異大的缺陷,是一種有效的癌癥基因表達測序分類新算法。
關鍵詞:RNA-Seq;LASSO回歸;特征提取;多層感知器神經(jīng)網(wǎng)絡;基因表達;TCGA數(shù)據(jù)庫
中圖分類號:TP3? ? ? ? 文獻標識碼:A
文章編號:1009-3044(2022)19-0091-03
轉錄組測序技術(RNA Sequencing,轉錄組測序技術)能夠對生物的轉錄本進行檢測,確定哪些變異在癌癥樣本中有表達,通過分析基因表達差異識別出變異基因或癌癥基因,在腫瘤疾病的診斷和治療起著重要作用,具有重要的科學意義與應用價值。但是,轉錄組測序技術可以在一次實驗中獲取大規(guī)模的基因表達譜數(shù)據(jù)[1],若要在海量的信息中識別疾病相關基因,使RNA-Seq技術在腫瘤疾病的診斷和治療中發(fā)揮重要作用,則要引用特征選取和機器學習的方法。為研究高效率、高準確率的基因分類算法,本文提出一種基于LASSO回歸和多層感知的癌組織RNA-Seq數(shù)據(jù)分類算法,在一次對癌癥樣本RNA-Seq測序后,可直接將結果進行識別、預測、分類。
為了解決高維基因數(shù)據(jù)的特征篩選和分類問題,1996年Robert Tibshirani[2]提出的LASSO回歸算法為基因特征數(shù)據(jù)的提取提供了技術支持,并逐漸應用到生物信息學領域。對于基因數(shù)據(jù)的特征篩選和分類問題,張靖等人[3]提出一種基于迭代Lasso的信息基因選擇方法,采用改進的Lasso方法進行冗余基因的剔除以獲得基因數(shù)量少且分類能力較強的信息基因子集,并使用支持向量機(SVM)、K近鄰(KNN)、決策樹C4. 5和隨機森林Random Forest4種分類器進行分類。張靖、張玉紅等人[4]提出K-split Lasso特征選擇方法,其基本思想是將數(shù)據(jù)集平均劃分為K份,分別使用Lasso方法對每份進行特征選擇,而后將選擇出來的每份特征子集合并,重新進行特征選擇,得到最終的特征基因,最后采用支持向量機進行分類。Ma[5]等人結合K-means和Lasso方法對基因表達譜數(shù)據(jù)進行特征選擇和預測模型構建,取得了較好的效果。
1本文算法原理
1.1 LASSO回歸原理
在樣本基因數(shù)據(jù)中引入的特征太多,主成分分析法選擇將一些原始數(shù)據(jù)丟失[6],而這些數(shù)據(jù)可能含有對樣本差異的重要信息,這就會對區(qū)分樣本類別的結果產(chǎn)生影響。采用LASSO回歸(Least Absolute Shrinkage And Selection Operator)更適用于處理一次RNA-Seq技術測序所產(chǎn)生的數(shù)據(jù),LASSO回歸通過參數(shù)縮減擬合廣義線性模型的同時進行變量篩選,從而達到降維和選取特征基因的目的[7]。這個方法能夠保留原有的基因特征屬性,選取關鍵特征,可直接用于特征建模分析。
以提取多種癌癥組織樣本特征為例:給定[n]個疾病樣本[{(X1,Y1),…,(Xn,Yn)}],自變量[X=(x1,x2,…,xn)T∈Rm*n]為基因數(shù)據(jù)矩陣,[xn∈Rm]為m維數(shù)據(jù)樣本,包含m個特征,響應變量[Y=(y1,y2,…,yn)T∈Rn],[Y]為疾病標簽,自變量[X]對響應變量[Y]進行線性回歸,約束[λ=(λ1,λ2,…λt)]不超過閾值[e]。
設本實驗目標函數(shù)為:
LASSO回歸優(yōu)化目標是令代價函數(shù)(cost function,或稱為損失函數(shù),lost function)最小,
[min L(λ)=12nj=1n(yj-λTxj)2+μj=1t|λj|subjecttoj=1t|λj|≤e](1)
n為樣本個數(shù),[μ]為正則化參數(shù),[t]為參數(shù)個數(shù)。隨著[μ]的增大,各變量的系數(shù)逐漸趨于零。
1.2 多層感知器
多層感知器(Muti-Layer Perception,MLP)是一種前饋式人工神經(jīng)網(wǎng)絡,是目前最成熟的人工神經(jīng)網(wǎng)絡之一。它由三層結構組成,分別是:輸入層、隱藏層和輸出層。MLP神經(jīng)網(wǎng)絡能學習和存儲大量輸入-輸出模式的映射關系,被廣泛應用于圖像,自然語言處理,生物信息領域識別、預測、分類[8]。
2 多層感知的癌組織RNA-Seq數(shù)據(jù)分類算法的構建
2.1 獲取數(shù)據(jù)集與基于R語言的數(shù)據(jù)處理
2.1.1 數(shù)據(jù)集的獲取與數(shù)據(jù)預處理
本次實驗樣本基因數(shù)據(jù)來源于TCGA數(shù)據(jù)庫,TCGA是關于癌癥方面的最大的公共數(shù)據(jù)集[9],為研究腫瘤學的人們提供了便捷的數(shù)據(jù)獲取平臺。本實驗使用3782個樣本進行建模,每個疾病樣本包含25190個基因,原始數(shù)據(jù)無法直接用于模型訓練,因此要進一步對數(shù)據(jù)進行處理。
從數(shù)據(jù)庫獲取到的基因數(shù)據(jù)集為COUNT矩陣,將COUNT矩陣導入R,把基因ID轉換為Gene symbol,去除重復的基因,選取表達量最大的基因,這些基因將用于做數(shù)據(jù)標準化。
2.1.2 數(shù)據(jù)編碼:One-Hot
本實驗序列的標簽將采用One-Hot的方法進行編碼。用LIHC、STAD、BRCA、DLBC、ESCA、GBM、OV、PAAD、LUAD、UCEC這10種癌癥基因數(shù)據(jù)進行分類,并將患病樣本所對應的疾病作標簽。
2.1.3 數(shù)據(jù)標準化
數(shù)據(jù)標準化的目的主要是消除測序數(shù)據(jù)的技術偏差[10],各個樣本基因數(shù)據(jù)間的測序深度和基因長度處于相同的水平,從而使我們得到具有生物學意義的基因表達量變化。本實驗則采用了文獻[11]的方法,使用基于R語言的voom函數(shù)對RNA-Seq基因數(shù)據(jù)標準化處理。
2.2 基于LASSO回歸的降維及特征提取的實現(xiàn)
LASSO回歸的核心思想是將不相關的特征系數(shù)變?yōu)榱悖瑥亩Y選出含有特征基因變量。具體實現(xiàn)如下:
(1)構造一個從200的-5次方到200的2次方的等比數(shù)列,這個等比數(shù)列的長度是200個元素,[λ]即這200個元素中不同的值。
(2)給定一個變量alphas,用于進行交叉驗證的正則化參數(shù)。令alpha=[λ],采用十折交叉驗證的方法找出最佳的alpha值,迭代1000次。
(3)調(diào)用最佳正則化參數(shù)下建立的模型系數(shù),輸出相關系數(shù)不為零的特征。
(4)記錄相關系數(shù)不為零的特征,用于構造新的數(shù)據(jù)集。
(5)劃分數(shù)據(jù)集,設定一個隨機種子,在任意帶有隨機性的類或函數(shù)里作為參數(shù)來控制隨機模式,得到新的數(shù)據(jù)集按7:3的比例劃分,得到比例為7:3的訓練集與測試集。
本實驗從25190個基因中提取到與標簽最相關的1414個特征基因及其表達量這些數(shù)據(jù)將用于模型訓練。
2.3 模型訓練
參數(shù)設置:實驗中MLP神經(jīng)網(wǎng)絡的激活函數(shù)設置為relu函數(shù),隱藏層設為3層,每一層隱藏層的神經(jīng)元設置為500,第一層隱藏層的學習率設置為0.1,第二、第三層的隱藏層學習率設置為0.2。
實驗環(huán)境:Intel CPU 3.20 GHz處理器,8 GB內(nèi)存的PC機,Windows 10操作系統(tǒng),PyCharm 2020.3.3開發(fā)環(huán)境。
①信息前向傳播
設[ol]=[(ol1,ol2,....,oln)T]為第[l]層的輸出,[l]=(1,2,3,4,5),n=(1,2,...,500)
當[l]=1時,
[oli]=[xi]? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? (2)
當[l]≥2時,
[ol=Wl*ol-1+bl]? ? ? ? ? ? ? ? ? ? ? ?(3)
當[l]=5時,此時為輸出層:使用多分類函數(shù)softmax計算得到輸出層的輸出:
[y=exp(o4)n=1500exp(o4n)]? ? ? ? ? ? ? ? ? ? ? ? ? ? ? [(4)]
隱藏層間的激活函數(shù)relu:
relu[(x)=max(0,x)]? ? ? ? ? ? ? ? ? ? ? ? ? ?(5)
②信息反向傳播
設代價函數(shù)(cost function)為[E],N為訓練樣本個數(shù):
[Etotal=12Ni=11||yi-xi||2]? ? ? ? ? ? ? ? ? ? ?[(6)]
優(yōu)化目標為確定W(權值)和b(偏置)使得損失函數(shù)[E]最小,采用梯度下降法更新參數(shù)的公式為:
[Wl=Wl-δNi=1N?EiWl]? ? ? ? ? ? ? ? ? ? ? ? ?[(7)]
[bl=bl-δNi=1N?Eibl]? ? ? ? ? ? ? ? ? ? ? ? ? ? ? [(8)]
式中:[δ]為學習速率,取值范圍(0,1]。
3 實驗結果與分析
本文采用BP網(wǎng)絡、CNN網(wǎng)絡做對比實驗用于驗證本文算法的優(yōu)勢。
3.1 實驗結果
3.2 實驗結果分析
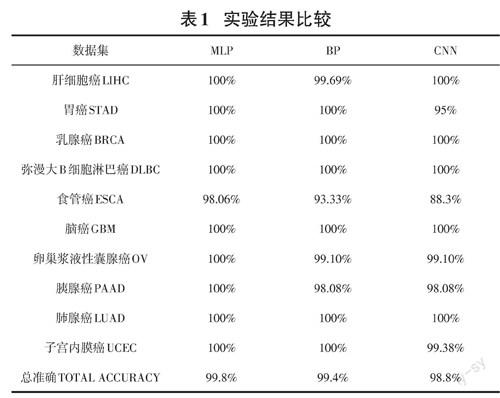
分別構建基于MLP、BP和CNN的分類模型,得到RNA-Seq基因樣本在3種模型下的識別準確率如表1所示。根據(jù)表1得知,在MLP模型中,準確率為99.8%,分類效果較為理想;在BP模型中,準確率為99.4%;在CNN模型中,準確率為98.8%,分類效果相對較差。根據(jù)上述的分類結果可知,MLP模型能夠使用多類別基因數(shù)據(jù)識別方式對RNA-Seq數(shù)據(jù)樣本進行有效區(qū)分,且效果最佳。
4 結束語
本文提出了一種基于LASSO回歸和MLP模型構建對多種癌組織樣本RNA-Seq基因序列的分類算法,本算法增加了訓練樣本數(shù)量,與其他神經(jīng)網(wǎng)絡的分類方法相比具有較好的分類效果,且優(yōu)于文獻[9]的分類算法準確99.3%。在LASSO回歸算法的基礎下,提取出樣本特征,為多層感知器提供了輸入數(shù)據(jù),增加了模型分類的準確率和進一步提高了泛化能力。基于LASSO回歸的多層感知器模型的識別的準確率為99.8%,符合多種癌癥RNA-Seq基因序列的分類需求,同時也為其他基因數(shù)據(jù)分類方法提供借鑒。
參考文獻:
[1] DERISI JL, IYER VR, BROWN PO. Exploring the metabolic and genetic control of gene expression on a genomic scale[J]. Science, 1997, 278(5338): 680-686.
[2] Tibshirani R. Regression shrinkage and selection via the lasso [J]. J Royal StatSocSer B Methodol, 1996, 58(1): 267-288.
[3] 張靖, 胡學鋼, 李培培, 等. 基于迭代Lasso的腫瘤分類信息基因選擇方法研究 [J]. 模式識別與人工智能, 2014,27(1): 49-59.
[4] 張靖, 胡學鋼, 張玉紅, 等. K-split Lasso: 有效的腫瘤特征基因選擇方法 [J]. 計算機科學與探索, 2012, 6(12): 1136-1143.
[5] MA SG, SONG X, HUANG J. Supervised group Lasso with applications to microarray data analysis [J].BMC Bioinform, 2007, 8: 60.
[6] 紀榮芳. 主成分分析法中數(shù)據(jù)處理方法的改進[J].山東科技大學學報(自然科學版), 2007,26(5): 95-98.
[7] 王福友,白冰,徐平峰.基于SIS的基因表達數(shù)據(jù)分析[J].長春工業(yè)大學學報, 2017, 38(5): 417-420.
[8] 張馳,郭媛,黎明.人工神經(jīng)網(wǎng)絡模型發(fā)展及應用綜述[J].計算機工程與應用,2021,57(11):57-69.
[9] 蔣文妍.基于RNA-Seq數(shù)據(jù)的癌癥標志物研究[D].天津:天津工業(yè)大學,2020.
[10] Conesa A,Madrigal P,Tarazona S,et al.Erratumto:a survey of best practices for RNA-Seq data analysis[J].Genome Biology,2016,17(1):181.
[11] YANG YH, DUDOIT S, LUU P, et al. Normalization for cDNAmicroarray data: a robust composite method addressing single and multiple slide systematic variation [J].Nucleic Acids Res,2002, 30(4): 15.
收稿日期:2022-03-20
基金項目:國家自然科學基金(61866006)
作者簡介:顏瀅(1997—),女,廣西靈山人,碩士,主要研究方向為生物信息計算、智能計算;李文敬(1964—),男,廣西南寧人,教授,主要研究方向為并行計算、智能計算;李松釗(1994—),男,廣西靈山人,碩士,主要研究方向為智能計算。
猜你喜歡
艦船科學技術(2022年15期)2022-09-14 09:21:50
電子制作(2019年15期)2019-08-27 01:12:00
電子制作(2018年19期)2018-11-14 02:37:08
自動化學報(2017年7期)2017-04-18 13:41:09
自動化學報(2017年11期)2017-04-04 02:52:58
中國生物醫(yī)學工程學報(2017年6期)2017-02-10 05:11:45
廣西科技大學學報(2016年1期)2016-06-22 13:10:38
計算機工程(2015年4期)2015-07-05 08:28:02
機電信息(2015年3期)2015-02-27 15:54:46
機械工程師(2015年10期)2015-02-02 01:13:49
