苯甲醛對甲苯歧化與烷基轉移催化劑的影響
2022-09-14 09:59:38閆乃鋒王宗霜
石油化工技術與經濟 2022年4期
關鍵詞:催化劑
閆乃鋒 王宗霜
(1 中海油惠州石化有限公司,廣東 惠州 516086;2 中國石油化工股份有限公司上海石油化工研究院,上海 201208)
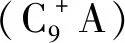
甲苯歧化與烷基轉移反應主要包含酸催化的烷基轉移反應以及芳烴側鏈加氫脫烷基反應,常用的催化劑為金屬改性的酸性分子篩雙功能催化劑[3-4]。在歧化與烷基轉移反應中,含氮化合物、硫化物、氯化物、烯烴類不飽和物等均能引起歧化與烷基轉移催化劑不同程度的失活。其中,堿性氮化物具有孤對電子,極性較大,極易吸附在催化劑的酸性中心上,形成穩定的絡合物,影響催化劑酸中心[5-6];硫化物和氯化物會與催化劑的金屬活性組分發生競爭吸附[7-10];烯烴類不飽和物則較易在催化劑表面縮合形成焦炭,而且還可能與其他組分發生烷基化反應,生成的副產物對芳烴產品的質量產生較大影響[11]。除此之外,CO可與催化劑中的金屬形成配合物,若氣相中CO含量較高,也會影響歧化催化劑,造成金屬活性中心中毒或者流失[12]。
隨著原料多元化以及劣質化的不斷升級,有機含氧化合物尤其是羰基類化合物經常出現在甲苯歧化與烷基轉移的原料中,而這類化合物對歧化催化劑的影響研究卻鮮有報道。文章以苯甲醛為探針分子,通過對不同含量的苯甲醛的反應原料在相同催化劑作用下反應性能的比較,考察了苯甲醛對催化劑性能的影響,并初步探索了其活性影響機理以及催化劑性能恢復方法。
1 實驗部分
1.1 實驗原料和裝置
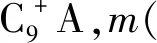
1.2 表征與分析方法
原料和液相產物分析采用Agilent 7890A氣相色譜儀,色譜柱為HP-FFAP柱,采用校正面積歸一法定量;苯產品純度采用確定餾分范圍的色譜歸一法計算得到;原料以及雜質定性分析采用Agilent 7890A/MSD 5977型氣相色譜-質譜聯用儀器(GC-MS),色譜柱為HP-1ms。
為表征甲苯和苯甲醛在歧化催化劑上吸脫附性能,采用英國海德分析公司(Hiden Analytical Ltd.)的智能重量分析儀(Intelligent Gravimetric Analyzer)來進行吸脫附行為研究。催化劑在573 K的真空條件下脫氣6 h,然后利用甲苯和苯甲醛蒸氣分別在298 K下對催化劑進行吸附,通過微量天平來記錄吸附過程中樣品的質量變化,最終獲得吸脫附平衡結果。
1.3 評價條件與數據處理
反應條件:壓力3.0 MPa,空速2.0 h-1,n(氫)∶n(烴)=2∶1,反應溫度370 ℃。
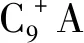
苯純度=液相產物中苯的質量/液相產物中(苯+環己烷+甲基環己烷+甲基環戊烷)的質量×100%
在實際工業生產中,因歧化反應中生成的副產物C6~C7非芳烴與苯的沸點接近,苯分餾塔無法通過精餾將苯和非芳烴分離,從而造成苯產品純度的降低。根據色譜分析結果來模擬計算工業上苯產品的純度(質量分數),影響苯產品純度的C6~C7非芳烴以環己烷、甲基環己烷和甲基環戊烷為主。
2 結果與討論
2.1 不同質量分數的苯甲醛對催化劑初始活性的影響
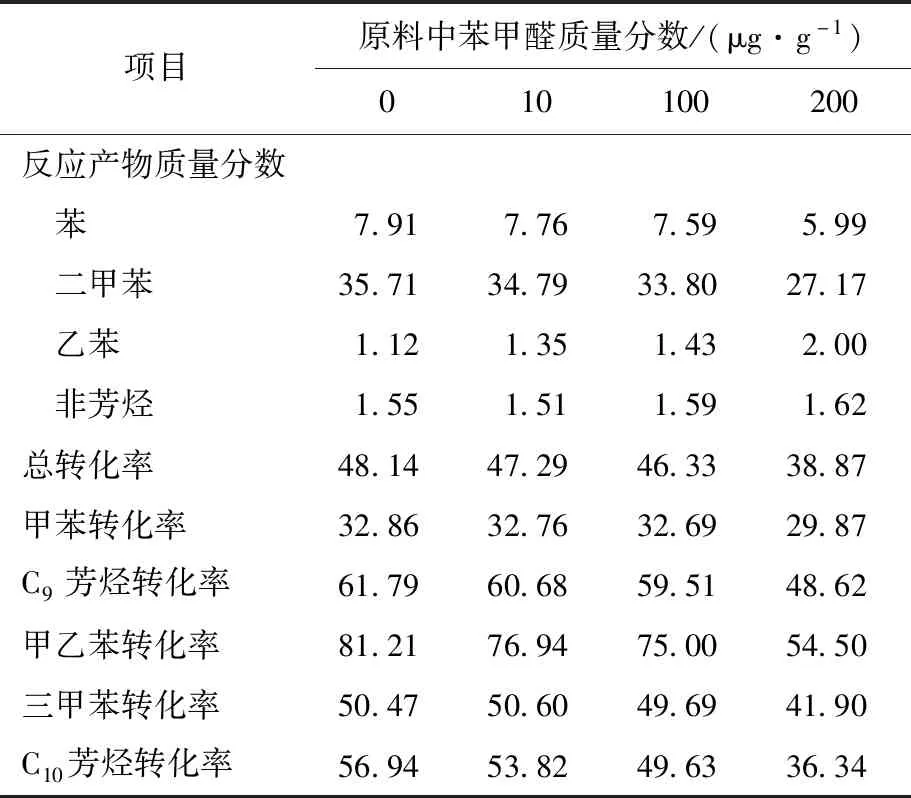
表1 苯甲醛對歧化催化劑性能的影響 %
2.2 苯甲醛對催化劑活性影響的可逆性研究
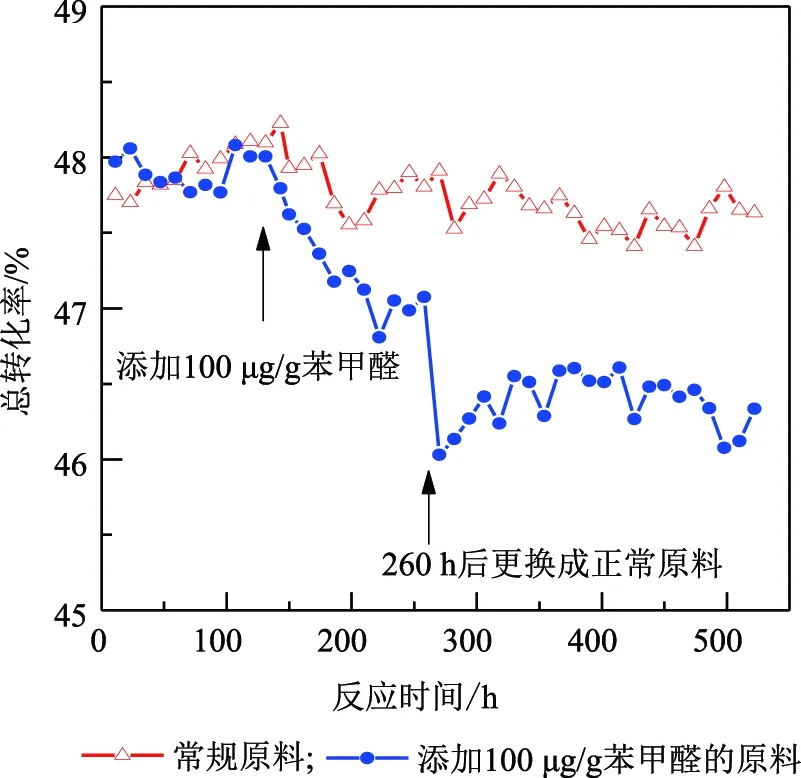
圖1 苯甲醛對催化劑反應性能的影響
由圖1可知:使用常規歧化原料,在500 h的反應時間內,反應總轉化率維持在48%左右。而在原料更換實驗中,常規歧化原料考評130 h后,將原料切換為含100 μg/g苯甲醛的歧化原料,催化劑反應總轉化率逐漸降低;考評至260 h時,反應轉化率已降低至46%。隨后,換回常規的歧化原料,反應活性趨于穩定,但僅得到部分恢復。這表明在短時間內,苯甲醛已經吸附在歧化催化劑的活性位上,僅僅通過撤出毒源,不能完全消除苯甲醛的影響,需借助其他手段來恢復催化劑活性。
為探究苯甲醛造成催化劑失活后反應性能的恢復情況,考評苯甲醛質量分數為150 μg/g的歧化原料,通過提高反應溫度和高溫脫附兩種方式來進行實驗,結果如圖2所示。
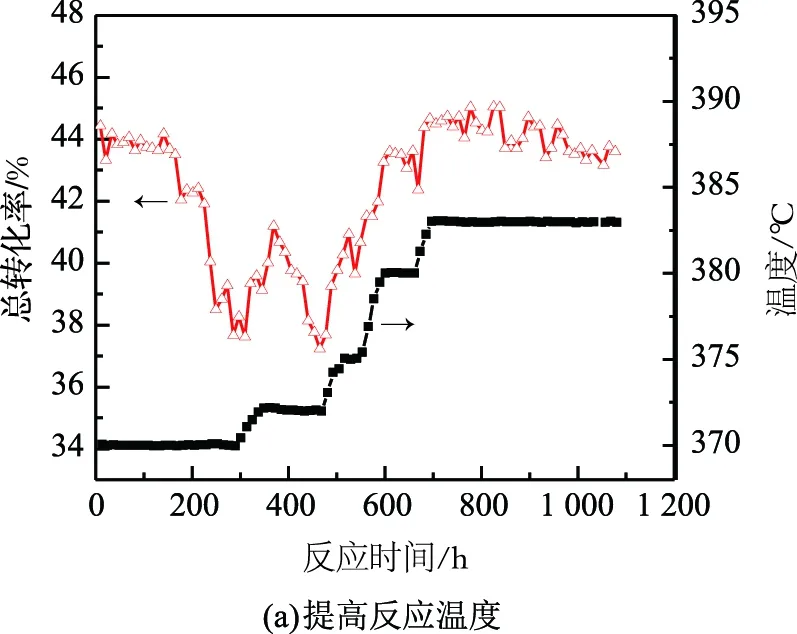
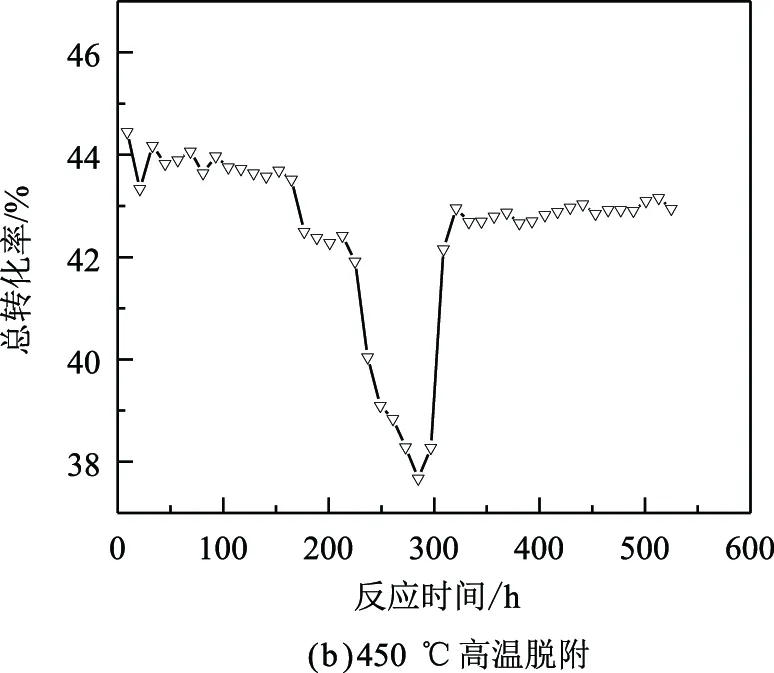
圖2 催化劑活性恢復實驗結果
圖2(a)反映了提溫實驗的反應性能變化趨勢。從圖2(a)中可以發現:在370 ℃反應溫度下,初期的催化劑轉化率緩慢降低,隨著反應的繼續,反應轉化率急劇降低。這是由于隨著反應的進行,苯甲醛在催化劑的活性位點上累積造成的結果。隨后,反應溫度由370 ℃逐漸升至383 ℃,反應轉化率已達到催化劑初期水平,383 ℃下運行近400 h,催化劑性能呈現緩慢降低的趨勢。由圖2(a)還可知:提高反應溫度,反應總轉化率有所恢復,但未撤出苯甲醛毒源,催化劑的穩定性不足。這表明提溫雖然能短時間提升催化劑活性,但失活趨勢仍然繼續。
圖2(b)反映了高溫脫附實驗的反應性能變化。由圖2(b)可以發現:催化劑經過450 ℃的停料氫氣脫附后,再將原料換為不含苯甲醛的常規歧化原料,轉化率有所上升,并逐漸接近初始活性,在200 h的反應時間內,活性相對較為穩定。這表明高溫氫氣脫附可以消除催化劑表面已吸附的苯甲醛,從而將催化劑表面的活性位點暴露,提高反應活性。因此,在短時間內,通過提溫和高溫脫附均可恢復催化劑部分性能,但需要及時撤出毒源,降低苯甲醛對催化劑性能的影響。
2.3 分析與討論
甲苯歧化與烷基轉移催化劑活性中心包含酸中心以及金屬活性中心,兩者協同催化,決定了催化劑的活性以及穩定性。Zaitan等[13]研究了甲苯和苯甲醛在Y沸石上吸附能力的差異。利用Langmuir吸附模型進行分析,在300 K下甲苯和苯甲醛在Y型沸石的最大吸附量分別為3.1 mmol/g和13.9 mmol/g,這表明在相同的反應條件下,苯甲醛比甲苯更易吸附在沸石分子篩上。Li等[14]在研究沸石催化劑苯甲醛催化氧化反應時發現,沸石載體具有強酸性位點,可以吸附極性較大的醛類化合物,且吸附強度要大于烴類化合物。由此可見,歧化反應中引入苯甲醛,可在酸中心上與芳烴發生競爭吸附,進而抑制催化劑烷基轉移活性。此外,馮愛虎等[15]在研究Beta分子篩以及Pd-MoO2/Beta催化劑上甲醛催化氧化反應過程時發現,甲醛不僅可以強吸附于Beta分子篩酸中心,而且可以吸附于PdOx金屬活性中心,通過酸中心和金屬活性中心的協同催化,顯著提升甲醛催化氧化活性。因此,可以推斷苯甲醛不僅影響歧化催化劑酸中心,而且同樣會影響其金屬加氫活性中心,降低催化劑加氫脫烷基活性,抑制芳環加氫副反應。
為佐證上述關于苯甲醛和甲苯對歧化催化劑吸附能力的差異,采用IGA智能重量分析儀表征其吸脫附行為,結果如圖3所示。從圖3可以清楚地觀察到,甲苯與苯甲醛分屬不同類型的吸脫附等溫線,苯甲醛的吸脫附等溫線存在明顯的回滯環。在相同的相對壓力下,苯甲醛吸附分支的吸附量與脫附分支的吸附量不能重合,說明苯甲醛與歧化催化劑產生了化學吸附,脫附較難,這與甲苯的吸脫附曲線形成鮮明的對比。從圖3中也可以發現:在相同壓力下,苯甲醛在歧化催化劑上的吸附量遠高于甲苯,這表明苯甲醛對歧化催化劑的吸附能力要高于甲苯,在歧化催化劑反應過程中優先吸附苯甲醛,從而對催化劑反應性能產生影響。
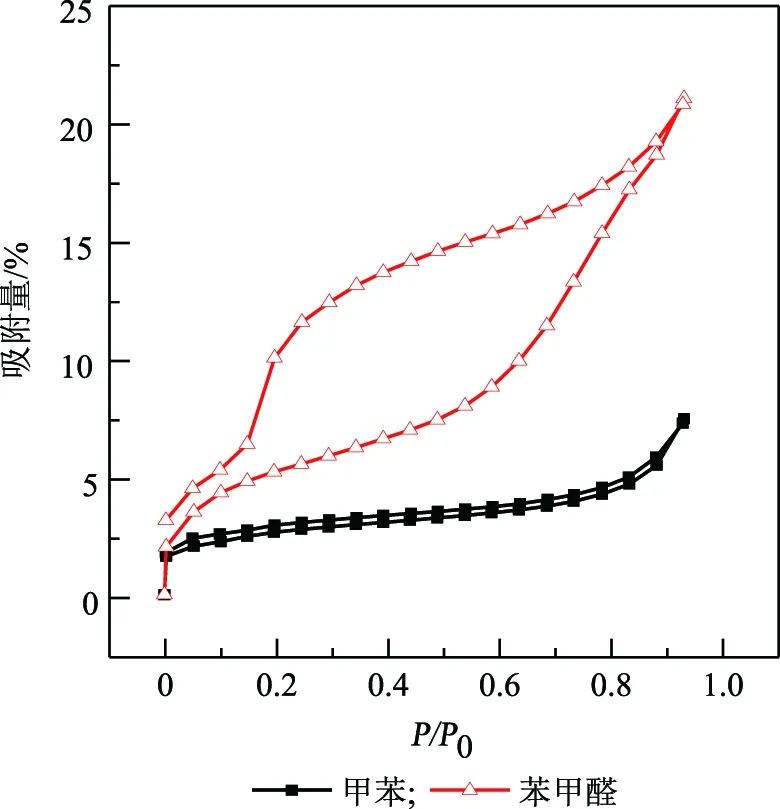
圖3 苯甲醛和甲苯在歧化催化劑上的吸脫附等溫線
結上所述,苯甲醛作為一種極性分子,在歧化催化劑酸性位上的吸附能力遠大于甲苯,與烷基轉移反應搶奪酸性位點,進而抑制催化劑的烷基轉移反應活性,使得各反應組分及反應總轉化率都有不同程度的降低。除此之外,苯甲醛同樣會影響歧化催化劑金屬活性中心,降低催化劑加氫脫烷基能力,使得甲乙苯的轉化率降幅明顯高于甲苯和三甲苯。與此同時,歧化原料中引入苯甲醛后,歧化催化劑中的金屬中心也會因為苯甲醛吸附,抑制了部分的苯環加氫副反應,隨著原料中苯甲醛質量分數的增加,苯產品純度也逐漸提高。因此,在甲苯歧化與烷基轉移反應過程中,苯甲醛作為一種新型毒物,應引起足夠重視,若遇到原料中苯甲醛超標情況,應及時停止進料,采取高溫脫附和更換原料的方法,恢復催化劑活性。
3 結論
(1)歧化原料中引入苯甲醛,可以持續降低催化劑烷基轉移以及加氫脫烷基活性。此外,苯甲醛的引入可以抑制苯環加氫副反應,使得苯產品純度提高。
(2)苯甲醛作為一種極性分子,在催化劑酸中心上的吸附能力遠大于甲苯,與烷基轉移反應搶奪酸性位點,降低烷基轉移反應活性。與此同時,苯甲醛同樣會吸附于金屬活性中心,抑制催化劑加氫脫烷基以及苯環加氫副反應,顯著降低甲乙苯轉化率,但是能有效提高苯產品純度。
(3)苯甲醛的吸附能力較強,撤出原料中的苯甲醛,催化劑活性僅有部分恢復,需通過提高反應溫度或者高溫脫附處理的方式,來減弱苯甲醛與催化劑之間的相互作用,短時間內可基本恢復催化劑的性能。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
