基于自噬與突觸可塑性探討電針治療缺血性腦卒中的機制
2022-09-16 10:15:58李煜超王燕鄒偉
上海針灸雜志 2022年9期
李煜超,王燕,鄒偉
(1.黑龍江中醫藥大學,哈爾濱 150040;2.寧夏醫科大學,銀川 750000;3.黑龍江中藥大學附屬第一醫院,哈爾濱 150040)
電針是中醫學臨床治療缺血性腦卒中(cerebral ischemic stroke, CIS)非常重要的治療手段。已有研究證明,電針可通過抑制炎性反應、腦水腫形成,減輕氧化應激損傷、細胞凋亡,促進神經與血管再生等多種途徑治療 CIS[1]。此外,有報道[2-3]顯示,電針可通過自噬和突觸可塑性途徑促進 CIS后神經恢復,但具體機制尚不完全清楚。筆者將從自噬途徑和突觸可塑性途徑兩方面對電針治療 CIS的相關機制進行綜述,并探討自噬與突觸可塑性在電針治療 CIS過程中的聯系,旨在進一步明晰電針治療CIS的內在機制。
1 電針治療CIS與自噬
1.1 CIS中影響自噬的因素
自噬是一種溶酶體降解途徑,可以清除受損細胞內細胞器和異常折疊的蛋白質及細胞內病原體,對真核細胞生存、分化、發育及內環境穩態至關重要[4]。在饑餓、缺氧、營養缺乏和感染等應激條件下,自噬可被激活為細胞的生存提供營養和能量[5]。自噬有 3種常見類型,即巨自噬、微自噬和伴侶介導的自噬,其中巨自噬與CIS的研究最為廣泛。巨自噬的發生發展(以下稱為自噬)有多個階段,包括自噬的啟動、囊泡成核、擴張和成熟以及自噬小體的融合和降解[6-7]。相關研究發現,在CIS的發生和發展過程中,自噬與多種細胞內生物學過程相互作用,包括自由基積累、線粒體功能失調和內質網應激(endoplasmic reticulum stress,ERS)的激活。這些過程與自噬在調節神經細胞死亡或存活方面有著復雜的聯系[8]。
CIS發生后,多種因素均可誘導自噬。當缺氧時,缺氧誘導因子(hypoxia inducible factor, HIF)被激活,HIF基因依賴性產物 B淋巴細胞瘤-2(B-cell lymphoma-2, Bcl-2)/腺病毒 E1B相互作用蛋白3(adenovirus E1B 19 kDa interacting protein 3,BNIP3)增加,BNIP3能競爭性結合Bcl-2,使 Beclin-1被釋放,從而啟動自噬[9]。缺血發生后,出現能量供應不足,單磷酸腺苷(adenosine monophosphate, AMP)與三磷酸腺苷(adenosine triphosphate, ATP)比值增大,AMP激酶(AMP-activated protein kinase, AMPK)被激活,AMPK可直接抑制雷帕霉素機能靶標蛋白復合物1(mechanistic target of rapamycin complex 1,mTORC1)并且磷酸化unc-51樣自噬激活激酶1(unc-51 like autophagy activating kinase 1, ULK1)來啟動自噬[10-11]。ERS出現時,內質網膜駐留蛋白肌醇依賴酶1α(inositol-requiring enzyme 1α, IRE1α)和RNA依賴性蛋白激酶(double-stranded RNA-dependent protein kinase, PKR)樣內質網激酶(PKR-like ER kinase, PEPK)被激活,它們可感知內質網管腔中未折疊蛋白的存在。激活的IRE1α通過激活c-Jun氨基末端激酶(c-Jun N-terminal kinase, JNK)介導的Bcl-2磷酸化導致Beclin-1/Bcl-2復合物的破壞,從而釋放自由的Beclin-1參與自噬體成核。PERK使真核起始因子 2α亞基(eukaryotic initiation factor 2α,eIF2α)磷酸化,磷酸化的 eIF2α通過上調自噬基因相關蛋白 12(autophagy-related protein 12, Atg12)的表達或促進 Atg5-Atg12-Atg16復合物形成參與自噬體膜的延伸。此外,ERS增加了細胞中鈣離子濃度,從而激活了AMPK途徑,緩解了mTOR對ULK1的抑制作用[12]。腦缺血性卒中后自噬過程見圖1。
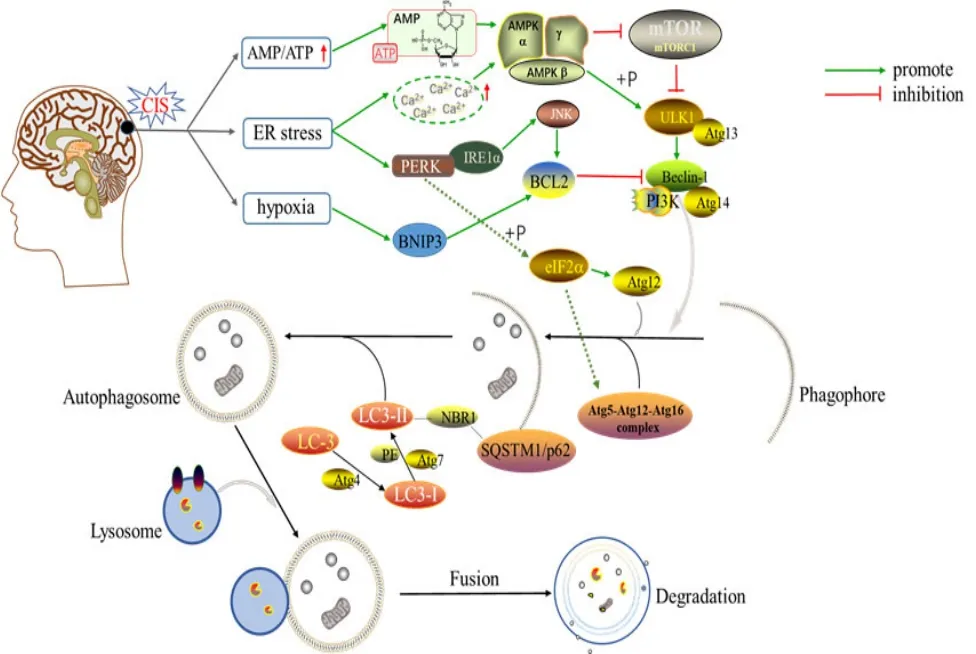
圖1 腦缺血性卒中后自噬過程
1.2 自噬在電針治療CIS的作用
電針治療 CIS對自噬的調節取決于時間節點[13],CIS發生2 h后,電針可促進自噬對抗神經損傷;而在24 h后,電針通過抑制自噬達到神經保護作用。此外,有學者[14]認為自噬在CIS過程中起雙重作用,適度的自噬對神經元起保護作用,而過度的自噬導致細胞損傷死亡。CHEN C等[15]研究發現,電針預處理通過抑制糖原合成酶激酶-3β(glycogen synthase kinase-3β, GSK3β),經 Wnt通路抑制自噬,從而誘導對腦缺血的耐受。黃亞光等[16]于術前5 d通過電針百會及雙側足三里、曲池等穴對右側大腦中動脈梗死(middle cerebral artery occlusion, MCAO)大鼠模型進行預處理,發現電針預處理治療 CIS的機制可能通過抑制自噬發揮神經保護作用。WANG M M等[17]觀察電針足三里和曲池穴對MCAO大鼠磷脂酰肌醇3-激酶(phosphatidylin-ositol-3-kinase, PI3K)/蛋白激酶B(protein kinase B, PKB或Akt)/mTOR通路的影響,于造模后24 h進行電針治療,發現電針可能通過調節PI3K/AKT/mTOR通路來抑制神經元自噬達到減少神經元死亡的作用。
有研究表明,減輕 ERS可有效緩解腦缺血再灌注損傷(cerebral ischemia reperfusion injury,CIRI)[18-19]。SUN X等[20]研究發現電針對CIRI損傷的神經保護主要是通過抑制ERS介導的自噬和細胞凋亡發揮作用。CIRI的發生促進了大量的促炎介質分泌,包括IL-6、IL-1β和TNF-α。有文獻證明,促炎性細胞因子可進一步促進嗜中性粒細胞和巨噬細胞浸潤到受損組織和炎癥反應中,導致炎癥反應發生和發展的惡性循環[21]。TING Z等[22]研究發現,電針治療CIS可以減輕缺血再灌注引起的炎癥反應。MATSUZAWAISHIMOTO Y等[23]認為自噬主要通過抑制炎癥小體,多蛋白復合物,以及通過自噬依賴性和非依賴性機制抑制I型干擾素(type I interferon, IFN-1)的產生來抑制炎癥。WANG X等[24]在研究心肌梗死炎癥與自噬時發現,自噬信號的早期增加和隨后的下降與促炎信號的出現保持一致,表明這兩種現象之間存在密切聯系。以上研究為電針調節自噬抑制缺血性腦卒中引起的炎癥提供了理論基礎。
自噬相關蛋白是哺乳動物細胞自噬程序正常執行所必需的。電針可以通過調節自噬相關蛋白來減少CIS的神經細胞損害。XU S Y[25]等研究發現,CIS早期給予電針治療,可以提高LC3-Ⅱ、Beclin1的表達促進自噬來改善MCAO大鼠神經功能障礙評分。另有研究[26]顯示,卒中后 24 h電針曲池和足三里穴可顯著提高mTORC1水平,從而導致ULK復合物失活,并抑制腦梗死后皮層中 Beclin1的磷酸化,提示電針通過抑制自噬小體的形成以及自噬而保護腦神經。CIS后線粒體受到破壞時,PTEN誘導的激酶1(PTEN-induced kinase 1,PINK1)會介導Parkin磷酸化,激活的Parkin介導線粒體外膜蛋白泛素化而促進線粒體自噬[27-29]。WANG H等[30]研究發現電針可改善自噬溶酶體途徑功能紊亂,通過PINK1/Parkin介導的線粒體自噬清除作用,減輕一氧化氮/氧化應激誘導的線粒體功能損傷,改善受損線粒體的積累,從而保護神經元免受CIRI。
2 突觸可塑性與CIS
2.1 CIS影響突觸可塑性的因素
突觸可塑性是指在內環境或外環境變化時,突觸在結構和功能上產生適應性改變的能力。在生理條件下,該過程與大腦發育,學習和記憶有關。在病理情況下,它參與腦損害的恢復[31]。LISMAN J[32]認為突觸可塑性的主要表現形式為短時程增強、早期長時程增強、晚期長時程增強、長時程抑制、距離依賴性縮放、穩態突觸縮放。
CIS后會導致突觸結構和功能的改變。谷氨酸是哺乳動物中樞神經系統的主要興奮性神經遞質,N-甲基-D-天冬氨酸(N-methyl-D-aspartate, NMDA)受體和 a-氨基-3-羥基-5-甲基-4-異惡唑丙酸(alphaamino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, AMPA)受體被認為是介導快速興奮性突觸傳遞的主要離子型谷氨酸受體[33]。AMPA受體在突觸后膜上穩定表達促進突觸傳遞的LTP,參與對大腦學習、記憶等認知功能的調節[34]。相關研究表明,慢性腦缺血可引起AMPA受體的谷氨酸受體2(glutamate receptor 2,GluR2)亞基內化增多,該亞基調控與鈣離子信號相關的突觸功能,導致鈣離子內流增加,使得細胞內一氧化氮水平增加,抑制線粒體的轉運功能,造成細胞內 ATP水平迅速下降,這一系列的調控機制最終損害神經突觸功能[35]。此外,該研究還提到腦缺血后蛋白激酶 C相互作用蛋白 1(protein interacting with C-kinase 1, PICK1)表達升高,PICK1是一種普遍存在的膜結合蛋白,它的PDZ結構域能與AMPA受體的GluR2亞基 C末端結合,使 GluR2內吞,導致突觸后膜上含GluR2亞單位的 AMPA受體減少,影響突觸傳遞的功能[36]。樹突棘是樹突上的小突起,是大多數興奮性突觸的突觸后位點,參與突觸信號的傳遞和整合,突觸的強度和活性與樹突棘的形態密切相關[37]。大腦皮質和海馬是機體最易受氧化應激和腦缺血缺氧損傷的區域,有學者研究了亞急性期到慢性期雙側感覺運動皮層和海馬樹突和樹突棘的動態變化,發現急性腦卒中后雙側感覺運動皮層和海馬的樹突和樹突棘均有不同程度的損傷,腦卒中后同側和對側感覺運動皮層樹突棘密度明顯降低[38-39]。
2.2 突觸可塑性在電針治療CIS的作用
潘愛環[40]觀察電針百會、大椎和腎俞穴聯合有氧運動對腦缺血模型大鼠認知功能、海馬組織形態學的影響,發現該療法能抑制PICKl的過表達,調節突觸后膜上AMPA受體的分布,從而改善慢性腦缺血導致的認知障礙。此外,電針可能通過抑制海馬CA1區嘌呤能受體 P2X配體門控離子通道 7(purinergic-receptor-P2X ligand-gated ion channel 7, P2X7R)的表達,促進突觸囊泡膜蛋白和突觸素的表達,改善突觸超微結構,促進突觸可塑性,進而改善大鼠的學習記憶功能[41]。
突觸后密度蛋白-95(postsynaptic density protein-95, PSD-95)是膜相關鳥苷酸激酶超家族的成員,是突觸后致密物中最豐富的支架蛋白,可連接谷氨酸受體、信號分子,以及其他結構蛋白,超過 95%的PSD-95表達定位于興奮性突觸[42]。Bé?QUE J C等[43]研究發現PSD-95的短期過表達通過選擇性促進AMPA受體介導的突觸傳遞和顯著增加這些突觸表達LTD的能力,從而明顯改變皮質間突觸的特性。突觸素(synaptophysin,SYP)是突觸前膜的特異性標志,可以促進突觸的形成,參與神經的修復、再生和突觸重塑[44-45]。郭斌等[46]研究發現,電針曲池和陽陵泉穴能夠升高大腦中SYP和PSD-95含量,促進了皮質突觸的重塑,從而改善中樞神經系統的運動功能。
突觸素P38是一種與突觸結構和功能密切相關的鈣結合蛋白,參與鈣離子依賴神經遞質乙酰膽堿和谷氨酸的釋放,還參與了神經元間信息的傳遞,對突觸傳遞效能的變化有明顯影響[47]。生長相關蛋白43(growth-associated protein-43, GAP-43)也被稱為神經調節蛋白,是伸長軸突和未成熟突觸終末的運動生長錐的主要成分[48]。在神經元的發育和再生過程中,GAP-43伴隨著軸突的生長在神經組織內大量合成[49]。有研究[50]表明,電針可以通過提高突觸素 P38和 GAP43在缺血中心區周圍皮層的表達,保護缺血性腦損傷。
3 電針治療CIS過程中自噬與突觸可塑性
近些年有較多學者分別研究了CIS中自噬及突觸可塑性對神經元的不同調節機制,發現自噬和突觸可塑性在缺血性腦卒中恢復過程中有重要作用。先前的研究提示自噬與突觸可塑性之間具有密切關系[51],而CIS后功能失調的自噬可能會引起一些突觸蛋白的紊亂,從而導致突觸功能障礙。有學者研究發現,自噬與突觸相關蛋白的表達呈負相關,應用自噬抑制劑后突觸相關蛋白的表達增加,而應用自噬激活劑后突觸相關蛋白的表達下降[52]。因此,自噬與突觸可塑性的聯系在電針治療CIS的過程中值得探究。
3.1 CIS后自噬與突觸可塑性的聯系
CIS后,由于腦組織缺血缺氧,增加了血管性癡呆(vascular dementia, VD)風險。有研究發現,自噬對VD大鼠海馬CA1區突觸可塑性相關蛋白GAP-43、SYP、PSD-95表達具有抑制作用,抑制自噬有利于VD大鼠海馬CA1區神經突觸重塑[53-54]。此外,自噬的誘導與AMPA受體亞基GluR1降解相關,在化學性LTD刺激神經元后,錐體神經元樹突和棘中的自噬體形成增加,使得 AMPA受體導向溶酶體從而降解[55],最終影響突觸可塑性。ZHANG X等[56]基于大鼠海馬神經元缺氧-葡萄糖剝奪(oxygen-glucose deprivation, OGD)模型和MCAO小鼠模型研究了缺氧缺血應激對自噬和突觸結構的影響,實驗發現缺血應激后出現功能性溶酶體儲存異常并伴有突觸功能障礙,具體表現為自噬短暫上調后出現溶酶體功能的紊亂,突觸蛋白1、突觸囊泡蛋白Ⅰ、TUBB3和突觸體相關蛋白 25等突觸功能相關蛋白表達的下調和蛋白質穩態破壞的相繼發生。此外,突觸中的自噬效應能降解SHANK3、PSD-95和PICK1等關鍵突觸后蛋白[57]。另有研究顯示,MCAO小鼠急性缺血后出現溶酶體相關膜蛋白 1的聚集和突觸超微結構的損傷,作者通過檢測突觸結構發現突觸功能障礙伴隨著溶酶體功能的改變,說明突觸蛋白動態轉換中的自噬依賴性損傷可能是突觸部位超微結構改變的原因,且溶酶體功能障礙可能是缺血應激條件下突觸功能障礙的分子基礎。
3.2 自噬與突觸可塑性在電針治療缺血性腦卒中的作用
腦源性神經營養因子(brain-derived neurotrophic factor, BDNF)作為一種治療腦損傷或神經退行性疾病的藥物已顯示出良好的前景,因為它可以改善認知功能,減少病理變化,防止神經元死亡[58]。BDNF作為突觸強度的活性依賴性修飾的關鍵介質,已知可增加突觸小泡的數量,并增強神經遞質的釋放。有研究指出,BDNF以其受體激酶 B(tropomysin related kinase B, TrkB)與PI3K途徑發生作用激活mTOR,從而抑制自噬來調節突觸可塑性[58]。ZHANG Y等[59]研究發現電針可以通過上調 BDNF的表達來抑制缺氧缺血大鼠海馬細胞死亡。另外,電針百會穴對MCAO模型大鼠進行干預,可以通過 BDNF/TrkB通路的表達促進腦缺血大鼠運動功能的恢復[60]。WANG H等[30]觀察了腦缺血再灌注過程中線粒體功能的損傷和受損線粒體的積累,發現電針對這兩種損傷均有改善,作用機制可能是通過PINK1/Parkin介導的線粒體自噬,提高自噬清除率從而改善受損線粒體的積累。CIS后出現線粒體功能障礙和線粒體累積,這種線粒體的異常導致突觸功能出現障礙[61],而線粒體自噬作為線粒體質量和數量的重要調節因素,可以有效改善線粒體功能[62]。通過線粒體自噬對線粒體的良性調節,可以支持突觸功能的恢復[63],為電針治療 CIS通過線粒體自噬改善突觸可塑性提供了理論依據。慢性腦缺血可引起突觸后膜上AMPA受體損傷,這種損傷可能是通過 PI3K-Akt-mTOR途徑介導自噬引起,應用 PI3K抑制劑能抑制自噬,部分恢復AMPA受體水平[55]。另有研究[17]發現,在CIS的治療過程中,電針可以通過 PI3K-Akt-mTOR通路抑制自噬,從而可能使 AMPA受體水平恢復,進一步調節突觸功能。
4 討論
自噬和突觸可塑性在 CIS恢復過程中有重要作用。目前自噬和突觸可塑性相關分子機制的研究尚處于探索階段,研究學者大多側重于對單個途徑的機制研究,而關于兩者或多個途徑之間聯系的報道較少,尤其是在電針治療CIS機制探討方面。本文從自噬和突觸可塑性兩方面分別綜述了電針治療CIS的不同機制,發現自噬在缺血早期可能對神經元起保護作用,而在再灌注期會導致細胞過度損傷而引起神經細胞死亡。電針治療作為缺血性腦卒中后神經功能恢復的重要干預手段,不僅可以促進自噬去除病理產物,還能抑制自噬抵抗不同疾病時期的細胞死亡,并且通過調節自噬抑制缺血性腦卒中引起的炎癥。自噬可以維持突觸的完整性和功能。此外,缺血性腦卒中后會出現突觸結構和功能的改變,電針干預后能促進突觸囊泡膜蛋白和突觸素的表達,改善突觸超微結構。
通過進一步探討自噬與突觸可塑性在電針治療CIS中的聯系,發現功能失調的自噬會引起一些突觸蛋白的紊亂,從而導致突觸功能障礙。自噬對突觸可塑性相關蛋白表達具有抑制作用,突觸中的自噬效應能降解關鍵突觸后蛋白。電針可以通過刺激 BDNF/TrkB的表達來抑制自噬從而調節突觸可塑性。在腦缺血損傷中,隨著活性氧、缺血/再灌注或毒素的刺激,導致線粒體和線粒體自噬功能的損傷。線粒體自噬途徑對維持突觸功能至關重要,而電針治療 CIS可通過調節線粒體自噬調節突觸可塑性。此外,電針可能抑制自噬提高AMPA受體的水平調節突觸功能。綜上所述,電針不僅可以通過分別調節自噬和突觸可塑性途徑促進 CIS后神經恢復,并且在自噬和突觸可塑性的關聯機制中發揮重要的調節作用。基于電針治療具有多靶點、多途徑的特點,從自噬和突觸可塑性及其聯系等多角度揭示電針對缺血性腦卒中的作用機理,對中醫學治療手段的現代化發展具有重要意義。
猜你喜歡
中華詩詞(2022年6期)2022-12-31 06:41:24
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
中國科技論壇(2017年7期)2017-07-25 08:49:53
汽車工程學報(2017年2期)2017-07-05 08:13:02
媽媽寶寶(2017年2期)2017-02-21 01:21:24
國際漢語學報(2016年1期)2017-01-20 08:21:20
