Gd對Mg-xGd-1Er-1Zn-0.6Zr合金顯微組織和腐蝕行為的影響
2022-09-19 10:26:48劉雄飛杜文博付軍健王云峰李淑波朱訓明王朝輝
材料工程 2022年9期
劉雄飛,杜文博*,付軍健,王云峰,李淑波,朱訓明,王朝輝
(1 北京工業大學 材料與制造學部,北京 100124;2 威海萬豐鎂業科技發展有限公司,山東 威海 264209)
鎂及其合金具有密度低、比強度與比剛度高、易切削加工以及可回收等優點,被譽為“21世紀綠色工程材料”,在汽車行業、3C電子產品和航空航天等領域有著廣泛的應用前景[1]。但純鎂的強度較低,其抗拉強度僅為40 MPa,無法作為結構材料獲得應用[2]。此外,由于鎂標準電極電位較低(-2.37 V),極易發生腐蝕,例如,Zhao等[3]發現純鎂在3.0%(質量分數,下同)NaCl溶液中的腐蝕速率達到2.7 mm/a。為進一步提高純鎂的力學性能和耐腐蝕性能,合金化是一條有效途徑,通過合金元素的固溶或時效析出第二相,不僅可以有效提高鎂合金的力學性能,同時也能改善鎂合金的耐腐蝕性能。例如,AZ91合金[4-5],因其沿晶界連續均勻分布的β-Mg17Al12相,使得其力學性能和耐腐蝕性能均獲得較大提升,鑄態抗拉強度達到159 MPa,在3.5%NaCl溶液中的腐蝕速率為1.1 mm/a。除Al,Zn等合金化元素外,添加RE元素也可有效提升鎂合金的力學性能和耐腐蝕性能。Li等[6]研究發現Mg-10Gd-4Y-0.3Zr合金峰時效后的抗拉強度達到325 MPa。Liang等[7]研究表明Mg-7Gd-3Y-0.4Zr合金經固溶處理后,在5.0%NaCl溶液中的腐蝕速率僅為0.5 mm/a。
近年來,含有長周期堆垛有序(long period stacking ordered,LPSO)相的稀土鎂合金因其優異的力學性能受到人們廣泛關注。Wang等[8]研究發現Mg-4Y-2Er-2Zn-0.6Zr合金固溶處理后,層片狀的14H-LPSO相轉變為塊狀的18R-LPSO相,其抗拉強度達到215 MPa,伸長率為21%。Ramezani等[9]研究表明Mg-8Gd-4Y-2Zn-0.6Zr合金經多向鍛造后,獲得超細晶組織及高體積分數的LPSO相,其抗拉強度達到581 MPa,伸長率為16%。雖然含LPSO相的稀土鎂合金力學性能因LPSO相得到顯著改善,但LPSO相對稀土鎂合金耐腐蝕性能的影響一直存在爭議。Zhang等[10]認為鑄態Mg-7Y-2Zn合金晶界處分布的連續塊狀LPSO相起到了腐蝕屏障的作用,合金在3.5%NaCl溶液中的腐蝕速率為2.3 mm/a。Bao等[11]認為鑄態Mg-9Y-3Zn-0.5Zr合金中致密的塊狀LPSO相能阻止腐蝕向基體擴張,使合金在3.5%NaCl溶液中的腐蝕速率控制在6.8 mm/a。然而,Zhang等[12]研究了鑄態Mg-11Gd-3Y-5Zn合金,發現晶界處不連續分布的LPSO相因與鎂基體電位差較大,充當強烈的陰極相加速腐蝕,在3.0%NaCl溶液中腐蝕速率達到756.8 mm/a。Bi等[13]研究發現Mg-12Dy-1Zn合金經過固溶處理后,晶粒內部析出大量層片狀的14H-LPSO相,在3.5%NaCl溶液中腐蝕速率達到267.7 mm/a。
為進一步分析LPSO相對稀土鎂合金的腐蝕行為的影響機制,本工作針對前期開發的Mg-Gd-Er-Zn-Zr高強鎂合金[14-15],通過改變Gd的含量,調控第二相數量和分布,研究第二相對合金腐蝕行為的影響規律,為闡明含LPSO相的Mg-RE合金的腐蝕機理提供理論依據。
1 實驗材料與方法
采用純Mg(99.99%),純Zn(99.99%),Mg-30%Gd,Mg-20%Er和Mg-30%Zr中間合金作為原材料。合金在井式電阻爐中進行熔煉,采用N2和SF6的混合氣體(體積比為20∶1)進行保護。熔體溫度為720 ℃時進行澆鑄,獲得鑄態合金。合金成分采用XRF-1800型X射線熒光光譜儀進行測試,結果如表1所示,三種合金中的實際Gd含量分別為6.66%,8.80%和10.76%,分別標記為合金A、合金B和合金C。采用D/MAX-3C型X射線衍射儀檢測合金和腐蝕產物的物相;采用Axio imager A2m金相顯微鏡(OM)和Quanta650型掃描電鏡(SEM)觀察試樣的顯微組織與表面形貌,并利用SEM設備上帶有的能譜儀(EDS)對試樣表面進行元素分析;采用Image J軟件分析試樣的顯微組織圖片,統計第二相體積分數和晶粒尺寸。

表1 三種合金的名義成分與實際成分(質量分數/%)Table 1 Nominal and actual compositions of three alloys (mass fraction/%)
在合金中間取樣,切成10 mm×10 mm×8 mm的試樣。將試樣懸掛浸泡在3.5%NaCl溶液中,溶液體積與試樣表面積比例為20∶1,浸泡不同時間后用鉻酸溶液清除腐蝕產物,計算試樣浸泡后的失重腐蝕速率。浸泡過程始終使用水浴保持溫度為25 ℃。
采用Autolab電化學工作站測試試樣的電化學性能,其中待測樣品為工作電極,鉑片為對電極(CE),與合金形成回路,飽和甘汞電極(SCE)為參比電極。在頻率為0.01 Hz~100 kHz范圍內進行電化學阻抗(EIS)測試,使用振幅為10 mV的正弦電壓,采用ZSimDemo3.30d軟件對電化學阻抗結果進行電路擬合。動電位極化測試的電壓范圍相對于開路電位±300 mV,測試是由負電位到正電位逐步掃描,掃描速度為0.5 mV/s。
2 實驗結果和分析
2.1 顯微組織
圖1為鑄態Mg-xGd-1Er-1Zn-0.6Zr合金的XRD圖譜,結果顯示這三種合金都是由α-Mg,Mg3Gd相和LPSO相組成。圖2為鑄態Mg-xGd-1Er-1Zn-0.6Zr合金的OM圖,由圖2可見,樹枝狀的黑色共晶相、灰色塊狀相和細小層狀相沿晶界分布,且隨Gd含量從7%增至11%,共晶相由不連續分布轉變為半連續分布,層狀相數量增多且貫穿晶粒內部,塊狀相逐漸消失。隨著Gd含量從7%增至11%,合金晶粒尺寸減小,從46.1 μm降至39.0 μm,但第二相的體積分數增加,共晶相的體積分數從1.9%增至5.2%,層狀相的體積分數從11.7%增至26.7%。

圖1 鑄態Mg-xGd-1Er-1Zn-0.6Zr合金的X射線衍射圖譜Fig.1 XRD patterns of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys

圖2 鑄態Mg-xGd-1Er-1Zn-0.6Zr合金光學顯微組織(a)合金A;(b)合金B;(c)合金CFig.2 Optical microstructures of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys(a)alloy A;(b)alloy B;(c)alloy C
圖3為鑄態Mg-xGd-1Er-1Zn-0.6Zr合金的SEM圖,圖中各點對應的EDS分析結果如表2所示。隨著Gd含量從7%增至11%,α-Mg內固溶的Gd元素含量從0.20%增至0.60%,Zn元素含量從0.02%增至0.12%,溶質原子溶入基體中能改善基體的表面電位且在基體表面形成致密的氧化膜[16]。合金內共晶相元素含量相同,(Mg,Zn)與(Gd,Er)元素的原子比均接近于3,結合XRD圖譜,可以確定共晶相為(Mg,Zn)3(Gd,Er)相。塊狀相中的元素成分接近Mg97Zn1(Gd,Er)2,與文獻報道[17]的LPSO結構成分范圍相近,結合XRD圖譜可以確定其為LPSO結構。層狀相主要含有Zn和RE(Gd,Er)元素,結合Jia等[14]的研究結果,確定其為幾個納米寬的LPSO結構。

圖3 鑄態Mg-xGd-1Er-1Zn-0.6Zr合金的SEM圖(a)合金A;(b)合金B;(c)合金CFig.3 SEM images of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys(a)alloy A;(b)alloy B;(c)alloy C
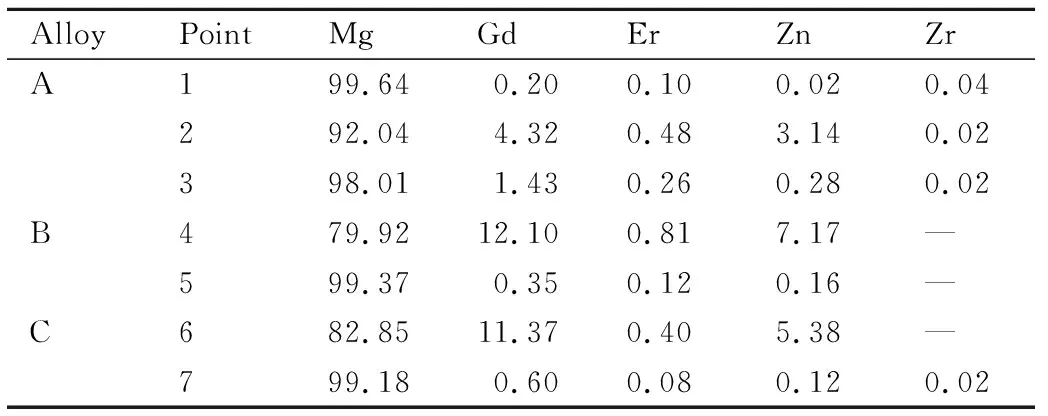
表2 圖3各點對應的EDS結果(原子分數/%)Table 2 EDS results of the various points shown in fig.3 (atom fraction/%)
2.2 電化學實驗
圖4為鑄態Mg-xGd-1Er-1Zn-0.6Zr合金在3.5%NaCl溶液中開路電位(OCP)的變化曲線。三種合金開路電位的變化都可分為3個階段,A階段表示開路電位迅速上升,B階段表示開路電位緩慢上升,C階段表示開路電位趨于穩定。開路電位的增加表明合金表面逐漸形成氧化膜,開路電位的降低表示合金表面的保護膜遭到破壞,開路電位保持相對穩定則代表腐蝕的進行與腐蝕產物的沉積達到了相對穩定的狀態[18]。如圖4所示,在A階段和B階段,三種合金的開路電位都隨著浸泡時間的延長逐漸上升,這是由于表面形成了保護氧化膜,隨著時間的延長,到達了C階段,腐蝕進行與腐蝕產物積累達到了動態平衡,這時開路電位保持相對穩定。開路電位圖中出現峰值的時間越早,代表局部腐蝕的潛伏期越短,表明局部腐蝕與腐蝕產物沉積之間的平衡越快[19],由圖4可見,合金A出現峰值的時間最長,為1609 s,合金C出現峰值的時間最短,為851 s。這表明合金C局部腐蝕的活化期較短,較容易被腐蝕,這可能是由于其存在較多的析出相。

圖4 鑄態Mg-xGd-1Er-1Zn-0.6Zr合金在3.5%NaCl溶液中的開路電位變化Fig.4 Variation of the open circuit potentials of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys during immersion in 3.5%NaCl solution
圖5為鑄態Mg-xGd-1Er-1Zn-0.6Zr合金在3.5%NaCl溶液中開路電位穩定后所測的電化學阻抗(EIS)曲線。根據開路電位測試,每個單獨的EIS測量都大約需要1800 s,而且整個測試過程都要處于開路電位穩定狀態。如圖5(a)所示,合金A的Nyquist圖由一個高頻容抗弧和一個低頻容抗弧組成,合金B和合金C的Nyquist圖都由一個高頻容抗弧和一個低頻感抗弧組成。高頻容抗弧反映材料基體的電荷轉移反應,低頻容抗弧反映電荷或物質通過表面氧化層的傳輸過程,低頻感抗弧的存在可歸因于局部腐蝕起始階段的腐蝕形核,可能是由于腐蝕介質中Cl-的存在使得表面膜更加活躍,低頻感抗弧的存在表明表面膜的保護能力不足,基體已經開始腐蝕[20]。高頻容抗弧越大,電荷穿過合金腐蝕產物膜與溶液之間的雙電層越困難,高頻容抗弧的直徑就等于工作電極的電荷轉移電阻,因而高頻容抗弧的大小可以反映耐蝕性的好壞[21],由Nyquist圖可見,合金A的高頻容抗弧最大,由此可判斷合金A耐蝕性最好。由Bode圖(圖5(b))可見,三種合金從高頻到低頻區域,阻抗值都一直上升,低頻區的模數值體現了合金的耐蝕性,結果顯示合金A在低頻區的模數值最大,表明其腐蝕傾向性最小。且由Bode圖中的相位變化可知,三種合金都在高頻區存在一個波峰,而在Bode圖中最大相位角越大,對應的頻率越低,代表合金的耐蝕性越好,結果顯示合金A的峰值最大,表明其耐腐蝕性能最好。采用電阻、電容和電感等電學元件構建等效電路對EIS圖譜進行進一步分析。圖6為鑄態Mg-xGd-1Er-1Zn-0.6Zr合金EIS阻抗譜的等效電路,圖6(a)用于擬合合金A的阻抗響應,圖6(b)用于擬合合金B和合金C的阻抗響應。圖中Rs代表溶液電阻;Rct代表電荷轉移電阻;CPEdl是恒相位元件,與電解質溶液和鎂基體界面的雙電層有關,用來代替理想的電容器來補償系統中的非均勻性[22];Rf表示薄膜電阻;Cf表示薄膜電容;L表示電感;RL表示電阻。Rct和CPEdl的并聯回路用來描述高頻電容弧,較高的Rct值代表鎂基體的溶解速率較低。Rf和Cf的并聯回路用來描述低頻電容弧,低頻電容弧的存在可歸因于合金表面氧化膜的存在[23]。RL和L的串聯回路用來描述低頻電感弧,低頻電感弧的出現是由開始發生局部腐蝕所引發,合金B和合金C存在低頻電感回路表示其耐蝕性較低。表3為利用ZsimDemo3.30d軟件進行擬合后得到的數據,其擬合誤差均在10-3數量級,表3顯示,三種合金的電荷轉移電阻分別是588.50,322.70,31.91 Ω·cm2,其數值越小,代表鎂基體溶解速率越大,合金C數值最小,代表其耐腐蝕性最差。

圖5 鑄態Mg-xGd-1Er-1Zn-0.6Zr合金在3.5%NaCl溶液中的電化學阻抗譜(a)Nyquist圖;(b)Bode圖Fig.5 Electrochemical impedance spectra of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys in 3.5%NaCl solution(a)Nyquist diagram;(b)Bode diagram

圖6 鑄態Mg-xGd-1Er-1Zn-0.6Zr合金電化學阻抗譜的等效電路(a)合金A;(b)合金B和合金CFig.6 Equivalent circuit models used for fitting the impedance spectra of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys(a)alloy A;(b)alloy B and alloy C
圖7為鑄態Mg-xGd-1Er-1Zn-0.6Zr合金的動態電位極化曲線,其中陽極分支(anodic branches)代表金屬陽極的溶解,陰極分支(cathodic branches)代表陰極析氫反應。由陽極分支可知,合金A更加陡峭,表示極化程度較大,耐蝕性較好[24]。由陰極分支可知,在電位比臨界點蝕電位更負時表現出線性Tafel特性,即存在Tafel線性區,且相比陽極分支更加陡峭,這表示陰極反應控制了腐蝕過程,且合金A的氫過電位最大,代表其耐蝕性最好。表4為Tafel外推法得到的鑄態Mg-xGd-1Er-1Zn-0.6Zr合金的電化學參數,由表4可見,三種合金的腐蝕電流密度分別為2.21×10-5,3.76×10-5,3.97 ×10-5A/cm2,合金A的腐蝕電流密度最小,且其極化電阻最大,約為2016.30 Ω,代表合金A的耐腐蝕性能最好。

表3 鑄態Mg-xGd-1Er-1Zn-0.6Zr合金電化學阻抗譜擬合結果Table 3 Fitting results of the EIS for as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys
2.3 腐蝕形貌
圖8為合金A在3.5%NaCl溶液中浸泡后帶有腐蝕產物的表面形貌圖。由圖8可見,浸泡1 h后,合金表面開始出現黃色膜層和黑色絲狀腐蝕產物,隨著浸泡時間的延長,表面的黑色絲狀腐蝕產物逐漸擴展,浸泡14 h后,合金表面已失去金屬光澤,表面覆蓋著灰白色的腐蝕產物層。
圖9為合金A在3.5%NaCl溶液中浸泡后帶有腐蝕產物的SEM圖,圖中各點對應的EDS分析結果如表5所示。浸泡1 h時,合金表面生成了針葉狀和花瓣狀的腐蝕產物,主要含有Mg,O和少量的稀土元素Gd和Er,且花瓣狀腐蝕產物中O元素含量較多,結合圖8可知,合金表面生成的黃色膜層是由針葉狀的腐蝕產物組成,黑色絲狀腐蝕產物是由花瓣狀產物連接形成。浸泡4 h后,針葉狀的腐蝕產物尺寸增加,花瓣狀的腐蝕產物逐漸堆積在合金表面,形成團絮狀,其中O和Mg元素的原子比接近于2,結合XRD圖譜,可確定其為Mg(OH)2。

圖7 鑄態Mg-xGd-1Er-1Zn-0.6Zr合金的動態電位極化曲線Fig.7 Potentiodynamic polarization curves of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys in 3.5%NaCl solution

表4 鑄態Mg-xGd-1Er-1Zn-0.6Zr合金的電化學參數Table 4 Electrochemical parameters of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys

圖8 合金A在3.5%NaCl溶液中浸泡不同時間后帶有腐蝕產物的表面形貌(a)0 h;(b)1 h;(c)4 h;(d)14 hFig.8 Surface morphologies of alloy A after immersion in 3.5%NaCl solution for different time with corrosion products(a)0 h;(b)1 h;(c)4 h;(d)14 h
圖10為鑄態Mg-xGd-1Er-1Zn-0.6Zr合金在3.5%NaCl溶液中浸泡8 h后的腐蝕產物XRD圖譜。結果表明,除了鎂基體以外,合金的腐蝕產物主要是由Mg(OH)2組成。從XRD圖譜中并沒有發現Zn,RE元素的氧化物以及β-(Mg,Zn)3(Gd,Er)和LPSO析出相,這說明合金在3.5%NaCl溶液中浸泡后,第二相自身沒有被腐蝕,但因其周圍的鎂基體被腐蝕后而發生脫落,部分進入溶液中,而殘留的第二相過少,未能被檢出[25-26]。且隨著Gd含量從7%增至11%,鎂基體的衍射峰強度降低,Mg(OH)2的衍射峰強度升高,說明合金C表面的腐蝕產物較多。

圖9 合金A在3.5%NaCl溶液中浸泡不同時間后帶有腐蝕產物的SEM圖(a)1 h;(b)4 hFig.9 SEM images of alloy A surfaces after immersion in 3.5%NaCl solution for different time with corrosion products(a)1 h;(b)4 h
圖11為鑄態Mg-xGd-1Er-1Zn-0.6Zr合金浸泡在3.5%NaCl溶液中去掉腐蝕產物后的SEM圖。由圖11可知,浸泡1 h后,晶界處最先開始腐蝕且出現寬度為5~27 μm的絲狀腐蝕坑,這是由于分布在晶界處的析出相因其電位較高充當陰極相,加速周圍鎂基體的腐蝕,因而晶界處的鎂基體最先開始腐蝕,當析出相周圍的鎂基體被腐蝕后,析出相發生脫落[27]。隨著浸泡時間的延長,腐蝕從晶界處向晶粒內部擴展,浸泡14 h后,合金A晶粒內部產生直徑約4 μm的點蝕坑,合金B表面出現直徑43~90 μm的巨大坑洞,合金C表面則出現數量較多的點蝕坑,直徑從2~15 μm不等。合金B表面的坑洞可能是由晶粒內部的點蝕坑擴展連接而形成。浸泡24 h后,腐蝕縱向擴展,留下較深的孔洞,合金A表面點蝕坑直徑增至15 μm,合金B表面出現大量纖維狀區域,合金C表面出現凹坑以及裂紋,纖維狀區域是由于析氫反應所產生的氣孔連接而形成[28],表明合金B析氫反應劇烈,腐蝕嚴重。

表5 圖9各點對應的EDS結果(原子分數/%)Table 5 EDS results of the various points shown in fig.9 (atom fraction/%)
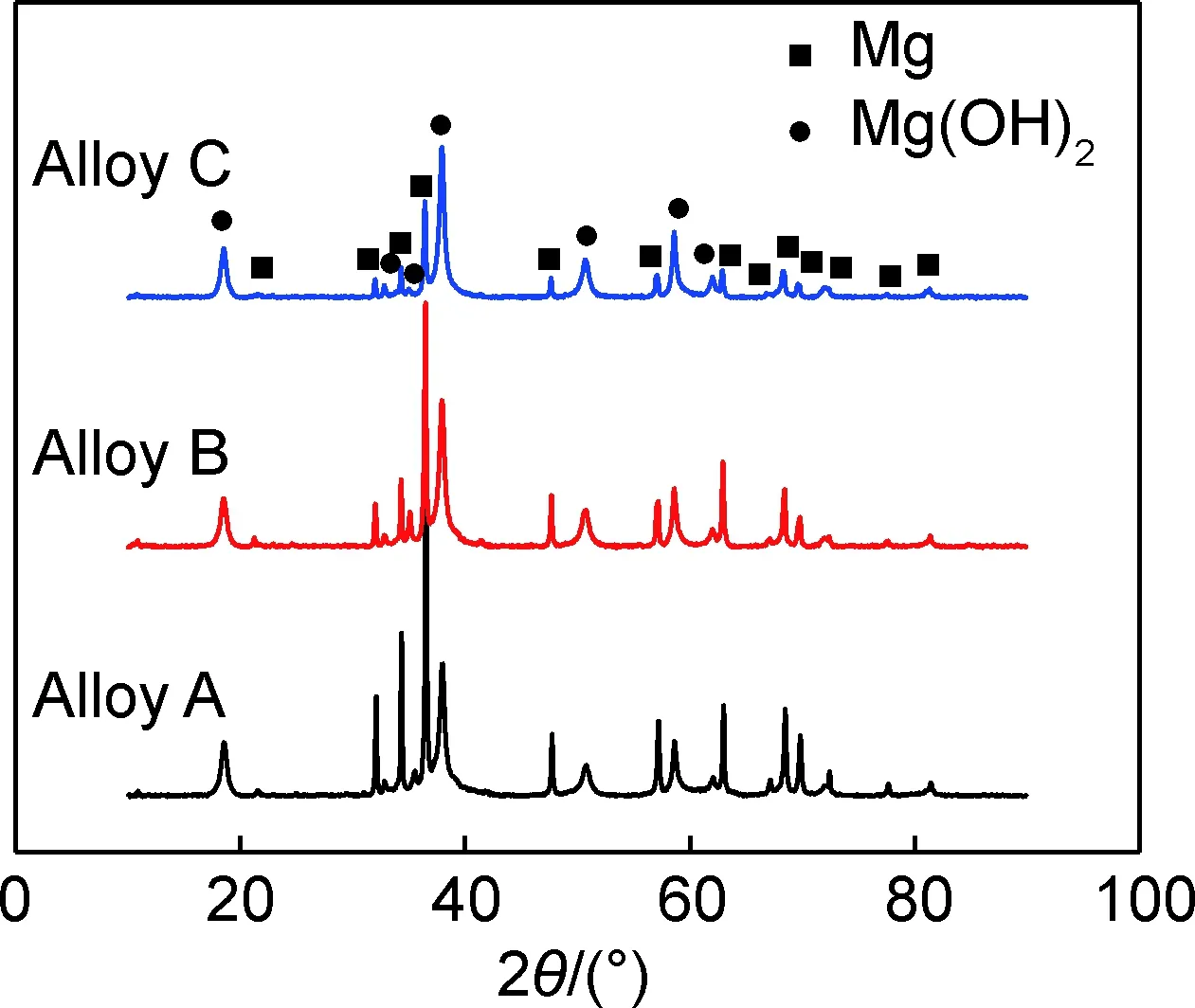
圖10 鑄態Mg-xGd-1Er-1Zn-0.6Zr合金在3.5%NaCl溶液中浸泡8 h后腐蝕產物的XRD譜圖Fig.10 XRD patterns of corrosion products of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys after immersion in 3.5%NaCl solution for 8 h

圖11 鑄態Mg-xGd-1Er-1Zn-0.6Zr合金在3.5%NaCl溶液中浸泡不同時間后去掉腐蝕產物的腐蝕形貌(a)合金A;(b)合金B;(c)合金C;(1)1 h;(2)14 h;(3)24 hFig.11 Morphologies of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys after immersion in 3.5%NaCl solution for different time without corrosion products (a)alloy A;(b)alloy B;(c)alloy C;(1)1 h;(2)14 h;(3)24 h
2.4 腐蝕速率
圖12為采用失重法測得的鑄態Mg-xGd-1Er-1Zn-0.6Zr合金的腐蝕速率。由圖12可知,合金的腐蝕速率都隨浸泡時間的延長而呈現增加的趨勢,經14 h浸泡后合金B的失重腐蝕速率更高。在浸泡初期,合金A的失重腐蝕速率更低,三種合金的腐蝕速率變化較緩慢,這是由于浸泡前期產生的腐蝕產物膜對合金有一定的保護作用。隨著浸泡時間的延長,Cl-不斷滲入腐蝕產物膜中,合金的失重腐蝕速率逐漸上升。浸泡14 h后,合金C的腐蝕速率增長減緩,低于合金B的腐蝕速率,這可能是因為合金C晶粒內部分布著的細小層狀LPSO相在一定程度上具有腐蝕屏障的作用,阻止腐蝕擴展,使得腐蝕速率增長減緩。在3.5%NaCl溶液浸泡實驗表明,鑄態合金的腐蝕速率順序如下:合金C>合金B>合金A。

圖12 鑄態Mg-xGd-1Er-1Zn-0.6Zr合金失重腐蝕速率Fig.12 Corrosion rate obtained by mass loss of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys
圖13為鑄態Mg-xGd-1Er-1Zn-0.6Zr合金的腐蝕機理示意圖。在腐蝕初期,晶界處分布著β-(Mg,Zn)3(Gd,Er)相、塊狀LPSO相和細小層片狀LPSO相,因其相對于鎂基體電位較高,充當陰極加速周圍鎂基體腐蝕,腐蝕最先開始于晶界附近,呈絲狀腐蝕的特征,部分第二相因周圍鎂基體腐蝕而脫落并溶入3.5%NaCl溶液中,殘留析出相繼續加速周圍鎂基體腐蝕。隨著浸泡時間的延長,合金表面開始形成氧化膜,根據圖10的XRD結果可知,氧化膜的主要成分是Mg(OH)2,而Mg(OH)2疏松多孔,無法對基體起到良好的保護作用。因而,Cl-不斷滲入氧化膜,加速腐蝕擴展,腐蝕從晶界處開始向晶粒內部蔓延,出現如圖11(c-2)所示大量的點蝕坑,點蝕坑逐漸擴展連接,形成了較大的坑洞。

圖13 鑄態Mg-xGd-1Er-1Zn-0.6Zr合金隨浸泡時間延長的腐蝕機理示意圖Fig.13 Schematic diagrams of the corrosion mechanism with immersion time of as-cast Mg-xGd-1Er-1Zn-0.6Zr alloys
合金A中第二相含量較少,微電偶腐蝕效應較弱,因而其腐蝕速率較低,合金B中第二相含量增加,微電偶腐蝕效應加強,導致腐蝕速率增大,且析氫反應劇烈,產生大量氣孔,形成纖維狀區域。合金C中β-(Mg,Zn)3(Gd,Er)相體積分數增至5.2%,并從沿晶界不連續分布轉變為半連續分布,層片狀LPSO相體積分數增至26.7%,并沿著晶界貫穿晶粒內部,β-(Mg,Zn)3(Gd,Er)相和層片狀LPSO相體積分數的增加,導致合金耐腐蝕性能下降,但大量細小層狀LPSO相能阻止腐蝕擴展,使得合金C在8~24 h內腐蝕速率增長減緩。
3 結論
(1)鑄態Mg-xGd-1Er-1Zn-0.6Zr合金中的析出相都沿著晶界分布,但當Gd含量從7%增至11%,合金析出相數量和分布都發生變化,β-(Mg,Zn)3(Gd,Er)相體積分數由1.9%增至5.2%,由不連續分布轉變為半連續分布。塊狀LPSO相逐漸消失,層片狀LPSO相體積分數由11.7%增至26.7%,且沿著晶界貫穿晶粒內部。
(2)開路電位測試表明,Gd含量較低的合金A到達峰值的時間最長,代表其局部腐蝕潛伏期較長,合金耐蝕性較好。電化學阻抗測試表明,Gd含量較高的合金C的高頻電容回路弧度最小,代表其耐蝕性較差。
(3)在3.5%NaCl溶液浸泡實驗表明,鑄態Mg-xGd-1Er-1Zn-0.6Zr合金的腐蝕速率順序如下:合金C>合金B>合金A。即隨著Gd含量的增加,合金耐蝕性下降,這主要歸因于第二相微電偶腐蝕效應和腐蝕屏障效應的共同作用。但大量細小的層狀LPSO相也能阻止腐蝕擴展,使得合金C在8~24 h腐蝕速率增長減緩。
