基于HPLC-Q-Exactive MS/MS的二味杜仲湯血漿成分研究
2022-09-20 07:55:10田顯庭張秀艷李春燕陸景坤薛培鳳
內蒙古醫科大學學報 2022年1期
田顯庭,董 馨,張秀艷,李春燕,陸景坤,薛培鳳*
(1.內蒙古醫科大學藥學院,內蒙古 呼和浩特 010059;2.呼和浩特市蒙醫中醫醫院藥劑科)
骨質疏松癥(OP)是一種全身性骨病,其特征是骨量減少和骨微結構嚴重受損,導致骨脆性增加,容易骨折[1]。骨質疏松癥引起的骨折已成為老年人死亡和致殘的主要原因之一。隨著我國人口老齡化的加劇,骨質疏松癥已成為我國面臨的重大公共衛生問題。中醫認為骨質疏松癥與骨萎縮相似,根據中醫理論,“腎主骨”“脾主肌”“氣血不通則痛”[2]。
目前治療骨質疏松癥的藥物以西藥為主,大部分為補鈣、維生素D、雙膦酸鹽類、激素類等,雖然這些藥物有固定的治療機制和靶點,具有顯著的療效,但是這些藥物副作用明顯,往往會對其他組織器官造成影響,因此亟待找到一種療效顯著同時副作用較小的藥物[2]。近年來蒙藥發展迅速,而其中二味杜仲湯就是一種蒙醫常用的治療骨科疾病的方劑,由杜仲和藍刺頭組成,具有壯骨、接骨療傷的功效[3]。此方使用多年,副作用較小,而且療效顯著,可以彌補西藥的不足。
此藥方又名塔布森-2,已有研究中多以體外質量控制和藥效研究為主,臨床研究表明此方對骨質疏松癥有顯著的療效,但是對于效應成分卻尚未闡述。藥物經過特定的傳輸途徑,入血、代謝、分布進而產生療效,因此,入血成分有可能是最終的效應成分,所以有必要研究血液中的成分,為進一步明確此方的藥效成分和代謝途徑提供科學依據[4]。
1 實驗材料
1.1 藥品與試劑
咖啡酸(批號:16081502,質量分數≥98%)、綠原酸(批號:SH17101905,質量分數≥98%)、異綠原酸C(批號:SH17120101,質量分數 ≥98%)、異綠原酸A(批號:SH17011305,質量分數≥98%)、原兒茶酸(批號:SH171208,質量分數≥98%)、龍膽酸(批號:SH17030706,質量分數≥98%)、綠原酸B(批號:SH18030501,質量分數≥98%)、阿魏酸(批號:SH17071213,質量分數≥98%)、表兒茶素(批號:16080706,質量分數 ≥98%)、隱綠原酸(批號:SH17103101,質量分數≥98%)、新綠原酸(批號:SH17100904,質量分數≥98%)、京尼平苷酸(批號:SH19011705,質量分數≥98%)、黃芩苷(批號:SH180206,質量分數≥98%)、紫云英苷(批號:SH17103002,質量分數≥98%)、松脂醇二葡萄糖苷(批號:SH18052807,質量分數≥98%)、桃葉珊瑚苷(批號:SH171110,質量分數≥98%)、去乙酰車葉草苷酸(批號:SH180206,質量分數≥98%),以上標品均購自北京賽百草有限公司;乙腈、甲醇(色譜純,美國Fisher公司);純凈水(杭州娃哈哈集團有限公司);甲酸(色譜純,天津永晟精細化工有限公司)。
本研究所用的杜仲、藍刺頭藥材經內蒙古醫科大學藥學院薛培鳳教授鑒定,分別為杜仲科植物杜仲(Eucommiau1moidesOliv.)的干燥樹皮、菊科植物驢欺口(EchinopslatifoliusTausch)的干燥花序。
1.2 實驗儀器
本實驗所用到儀器及具體信息見表1。
表1 實驗儀器Tab.1 Experimental equipments
1.3 實驗動物
Wistar雄性大鼠(購自內蒙古大學實驗動物研究中心,SPF級),體質量(240±20)g,許可號:SCXK(內蒙古)2016-01。動物在內蒙古醫科大學實驗動物研究中心常規飼養。適應性喂養7d,給予標準飼料和飲用水,保持(24±2)℃的室溫、(55±5)℃的相對濕度和12 h的暗光周期。實驗前禁食12 h,正常飲水。
2 實驗方法
2.1 色譜條件
色譜柱:ACE Excel 3 C18-PFP(3μm,100 mm×3 mm);流動相中有機相(A)為0.2%甲酸甲醇,水相(B)為含0.2%甲酸-超純水,梯度洗脫程序為:0~0.8 min,2%~5%(A);0.8~1.7 min,5%~10%(A);1.7~2.5 min,10%~18%(A);2.5~3.3 min,18%~23%(A);3.3~4.2 min,23%~28%(A);4.2~5.8 min,28%~33%(A);5.8~9.2 min,33%~43%(A);9.2~11.2 min,43%~46%(A);11.2~14.3 min,46%~60%(A);14.3~16 min,60%~65%(A);16~17.7 min,65%~68%(A);17.7~19.2 min,68%~75%(A);19.2~20 min,75%~80%(A);20~20.8 min,80%~2%(A);20.8~24 min,2%(A),流速為0.3 mL/min,4℃自動進樣,進樣量為5μL,柱溫30℃。
2.2 質譜條件
采用電噴霧離子源(ESI)負離子檢測,全掃描(full-scan)模式下獲取質譜信息。由于黃酮及酚酸類成分在負離子模式(ESI-)下離子化效果較好,且酚酸類成分在正離子模式下響應較差,因此本文采用負離子模式進行檢測。質譜條件如下:①鞘氣流速(Sheath gas flow rate):35 L/min;②輔助氣流速(Aux gas flow rate):10 L/min;③噴霧電壓(Spray voltage):2.8 kV;④毛細管離子傳輸管溫度(Capillary temperature):350℃;⑤S-lens電壓(S-lens RF level):50 kV;⑥加熱溫度(Aux gas heater temperature):150℃。
2.3 原型成分數據處理
首先建立EDD化學成分數據庫,通過中國知網、PubMed、ETCM、TCMSP、SymMap等網絡數據庫或平臺檢索TFES中各單味藥材中的化學成分,建立一個包含化合物名稱、化學式和分子量的數據庫。將負離子模式下檢測到的HPLC-(-)ESI-MS譜導入Xcalibur 3.0軟件進行質譜數據處理,得到質荷比(m/z)、保留時間(Rt)等信息,通過高分辨質譜計算出的精確分子量,結合已經完善的數據庫信息和對照品的比對,篩選出體內原型成分。質譜偏差范圍δ ≤5×10-6。
2.4 體內代謝數據處理
將含藥血漿和空白血漿的原始譜圖導入到Compound discover3.0軟件,并將EDD的體外化合物列表以mol格式導入到數據庫中,保留時間誤差設為0.3 min,質核比誤差為5 ppm,最多反應步數為3。在體內代謝模式下計算每個化合物的所有I相和II相代謝產物,推測各類化合物可能的代謝過程。
2.5 對照品溶液及灌胃藥液的制備
2.5.1 對照品溶液的制備 精確稱取各對照品適量,置于10 mL容量瓶中,加入甲醇稀釋至刻度,使體積恒定。制備每種濃度為1 mg/mL的化合物儲備溶液。所有儲備溶液儲存在4℃的冰箱中,以備日后使用。
2.5.2 灌胃藥液的制備 精密稱取EDD藥材細粉50 g(杜仲、藍刺頭各25 g),置于1 L圓底燒瓶中,加入500 mL蒸餾水,浸泡30 min,100℃,回流提取2次,每次30 min,用180目篩過濾,兩次藥液混合,濃縮后冷凍干燥,以獲得凍干粉供日后使用。使用時用適量蒸餾水溶解,使凍干粉含量為20 mg/mL(相當于EDD生藥量83.3 mg/mL)。凍干粉置于4℃冰箱保存備用。
2.6 血漿樣品的制備
取雄性Wistar大鼠10只,其中空白組3只,給藥組7只。空白組給予5 mL蒸餾水,給藥組給予5 mL的藥物溶液(相當于3.33 g/kg的EDD凍干粉)。給藥組大鼠給藥15、30、60、120、240、360 min后,腹腔注射10%水合氯醛(0.3 mL/100 g)用于麻醉,從腹主動脈采血,置于肝素化采血管中,在3500r· min-1下離心10 min,取上層血漿并放置在-20℃下,保存在冰箱中以備日后使用。
2.7 血漿樣品的處理
取各時間點血漿200μL,加入3倍量(含0.15%甲酸)乙腈,旋渦并混合1 min,在4℃下以13000r· min-1離心血漿樣品10 min以沉淀蛋白質,取上清液,并在室溫下用氮氣吹干上清液,將殘余物溶解在200μL 50%色譜甲醇中,旋渦1 min,在13000r· min-1和4℃下離心10 min,并取上清液供使用。
2.8 樣品前處理優化
2.8.1 給藥劑量優化 取2.5.2項下凍干粉適量,分別對大鼠進行灌胃給藥(劑量分別為1.67 g/kg、3.33 g/kg、5.00 g/kg、6.67 g/kg)。所有大鼠以10%水合氯醛(0.3 mL/100 g)麻醉。將采集的全血置于5 mL肝素化EP管中,3500r·min-1離心10 min,分離血漿。用乙腈沉淀蛋白法進行處理,并進行質譜分析。其中1.67 g/kg(臨床等效劑量的5倍)下檢測到的成分數目較少,5.00 g/kg和6.67 g/kg(臨床等效劑量的15倍和20倍)無明顯差別,3.33 g/kg(臨床等效劑量的10倍)下檢測到的成分數目最多,且基線最好,故選擇3.33 g/kg為最終給藥劑量(見圖1)。
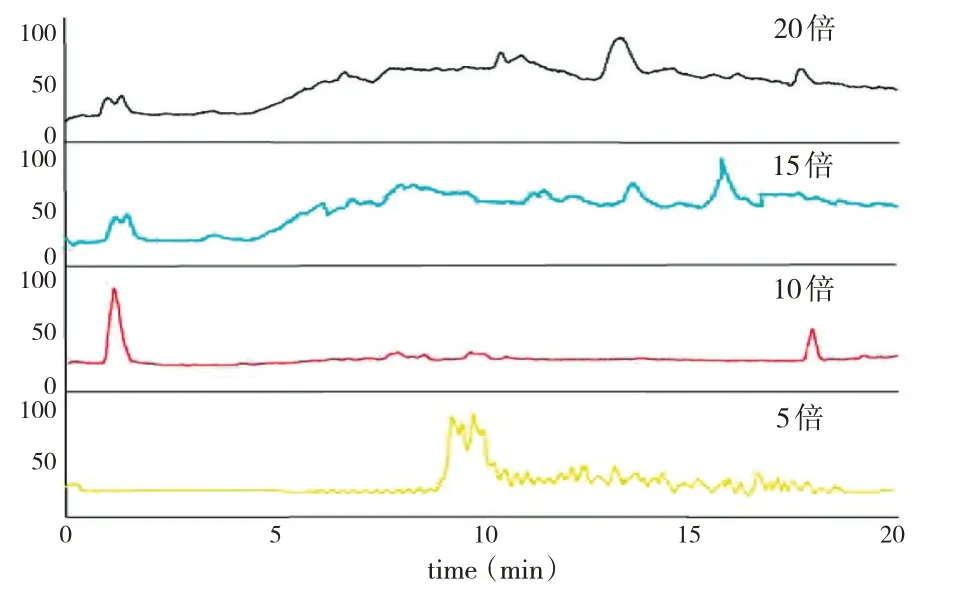
圖1 不同給藥劑量的大鼠血漿總離子流圖Fig.1 Total ion current diagrams of rat plasma in different doses
2.8.2 采血時間的優選對給藥后不同采血時間點的血樣進行預處理,通過進樣采集質譜數據。綜合比較15、30、60、120、240、360 min的譜圖,可以發現240 min后譜圖中TFES的原型成分已基本消除,因此在給藥后15、30、60、120 min收集大鼠。分析用血樣,其中120 min時檢測到的成分最多且峰形好。
2.8.3 含藥血漿沉淀蛋白方法優選為了使待測樣品測得的圖譜整體響應值更好,峰數目更多,峰強度更高,實驗前期對血樣的處理方法進行了優化。我們考察了不同蛋白沉淀劑對整體圖譜中待測成分的影響。采用乙腈、甲醇分別以不同比例(1∶2、1∶3、1∶4)作為沉淀劑,結果發現乙腈按照1∶3的比例作為沉淀劑時圖譜整體峰多,內源性干擾較少,且多數入血成分富集程度較高,響應值較好(見圖2)。
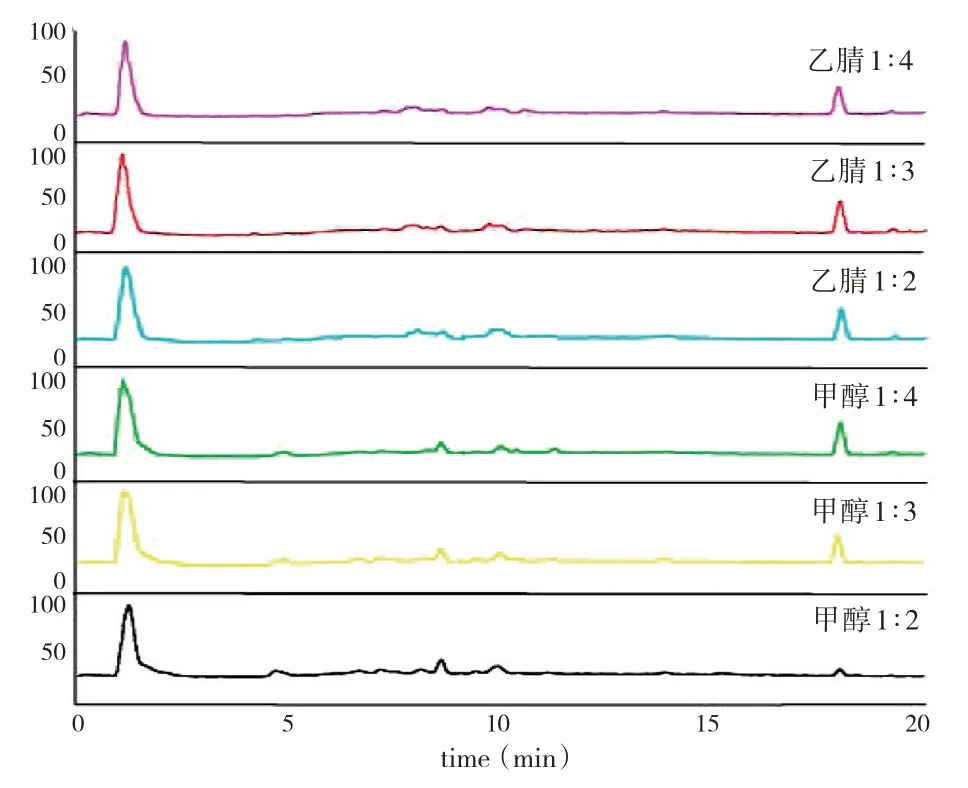
圖2 不同處理方式的大鼠血漿總離子流圖Fig.2 Total ion current diagrams of rat plasma with different treatments
2.8.4 色譜條件優化 本實驗前期比較了流動相水相中甲酸的含量(0%、0.1%、0.2%、0.3%、0.4%、0.5%,v/v)對入血成分分離度和靈敏度的影響,發現當流動相中不加甲酸,以綠原酸、咖啡酸為代表的酚酸成分嚴重拖尾,低濃度條件下難以基線分離。隨著甲酸濃度的增加,酚酸類成分拖尾現象顯著改善,但同時黃酮類成分呈現峰面積先增加后減小的趨勢,酚酸類成分峰面積明顯減小。最終確定以加入體積分數0.2%甲酸超純水作為水相,方劑血漿圖譜整體性較好。
2.9 二味杜仲湯入血原型成分分析
通過建立EDD化學成分數據庫,共收集了組方藥材中的原型成分246個。通過全面分析含藥血漿、空白血漿、EDD方劑、杜仲藥材、藍刺頭藥材的質譜圖,以高分辨質譜計算的精確分子量搜索各圖譜準分子離子峰,并通過保留時間比對確認各圖譜的共有峰,同時結合二級質譜碎片信息確認藥材吸收入血的原型成分。空白血漿中出現共有色譜峰,視為體內的內源性成分,暫不做吸收入血成分分析。大鼠給藥2 h后的總離子流圖見圖3。
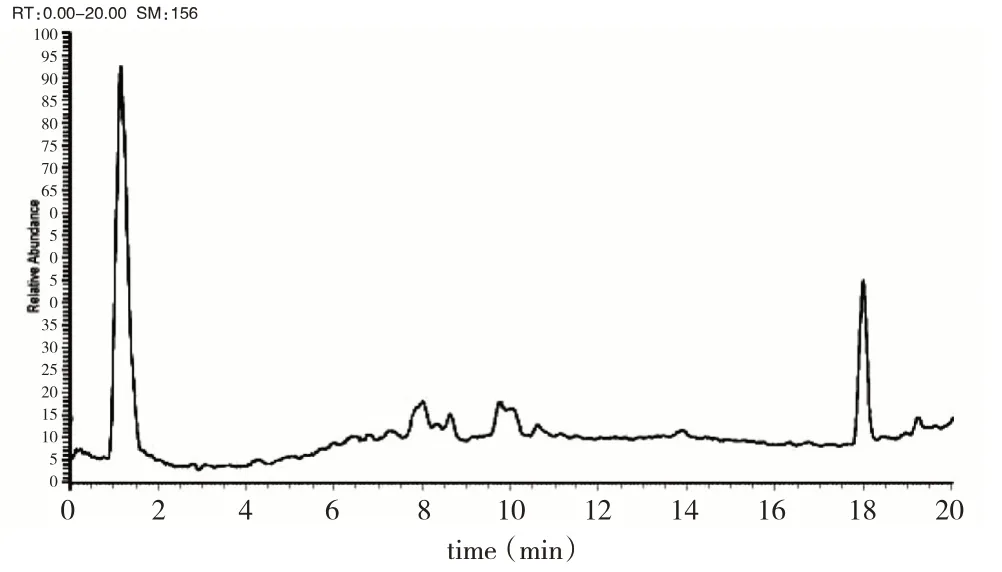
圖3 給藥2 h后大鼠血漿的總離子流圖Fig.3 Total ion current diagram of rat plasma two hours after administration
3 結果
通過全面分析含藥血漿、空白血漿、EDD方劑、杜仲藥材、藍刺頭藥材的質譜圖,使用高分辨質譜計算的精確分子量搜索各圖譜準分子離子峰,并通過保留時間比對確認各圖譜的共有峰,同時結合二級質譜碎片信息確認藥材吸收入血的原型成分。此外將含藥血漿和空白血漿原始圖譜導入Compound discover 3.0軟件,并將EDD的體外化合物以mol格式導入軟件,計算體內代謝模式下各化合物的所有Ⅰ、Ⅱ相代謝產物,獲得化合物的保留時間、質量核比、反應類型、匹配度等信息,結合軟件分析結果和參考文獻推測可能的代謝物。共鑒定出43個入血移行成分,其中有29個原型成分,14個代謝成分。結果見表2、表3。

表2 EDD血漿中原型成分的鑒定Tab.2 Identification of prototype components in EDD plasma

表3 EDD代謝產物鑒定Tab.3 Identification of EDD metabolites
3.1 黃酮類化合物代謝產物
M1保留時間17.973 min,分子式C17H18O7,誤差0.531,在負離子模式下準分子離子峰為333.10525,比貫眾苷準分子離子峰315.08631多18 Da(H2O)。根據軟件分析為貫眾苷的水合反應,因此,化合物初步判斷為貫眾苷的水合產物。
M2保留時間19.52 min,分子式為C19H22O6,誤差0.955,在負離子模式下準分子離子峰為345.14164,比貫眾苷準分子離子峰315.08631多30 Da(C2H6)。根據軟件分析,可能存在還原反應、水合反應、乙酰化反應,最終生成此化合物。
M3保留時間10.7 min,分子式為C18H16O5,誤差0.76,在負離子模式下準分子離子峰為311.09977,比貫眾苷準分子離子峰315.08631少4 Da(+C-O)。根據軟件分析,可能存在甲基化反應,脫水反應最終生成此化合物。代謝規律推導圖見圖4。
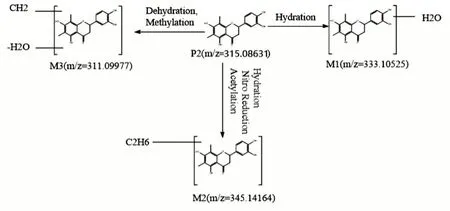
圖4 貫眾苷在大鼠體內代謝規律圖Fig.4 Metabolism of guanzhong glycoside in rats
3.2 苯丙素類化合物代謝產物
M6保留時間12.22 min,分子式為C9H8O3,誤差0.819,在負離子模式下準分子離子峰為163.04734,比咖啡酸準分子離子峰179.04226少16 Da(-O)。根據軟件分析,可能存在脫水反應和還原反應,最終生成此化合物。
M7保留時間11.14 min,分子式為C11H11NO3,誤差1.39,在負離子模式下準分子離子峰為204.07389,比咖啡酸準分子離子峰179.04226多25 Da(-O+C2H3N)。根據軟件分析結果,可能存在硝基還原反應,甘氨酸結合反應,最終生成此化合物。
M8保留時間9.73 min,分子式為C9H10O3,誤差為0.529,在負離子模式下準分子離子峰為165.06299,比咖啡酸準分子離子峰179.04226少14 Da(-O+2H)。根據軟件分析結果,可能存在還原反應,羥基化反應,最終生成此化合物。
M9保留時間7.17 min,分子式為C14H12N2O4,誤差為0.047,在負離子模式下準分子離子峰為271.07971,比咖啡酸準分子離子峰179.04226多92 Da(+C5H4N2)。根據軟件分析結果,可能存在脫水反應,谷氨酰胺結合反應,最終生成此化合物。代謝規律推導圖(見圖5)。
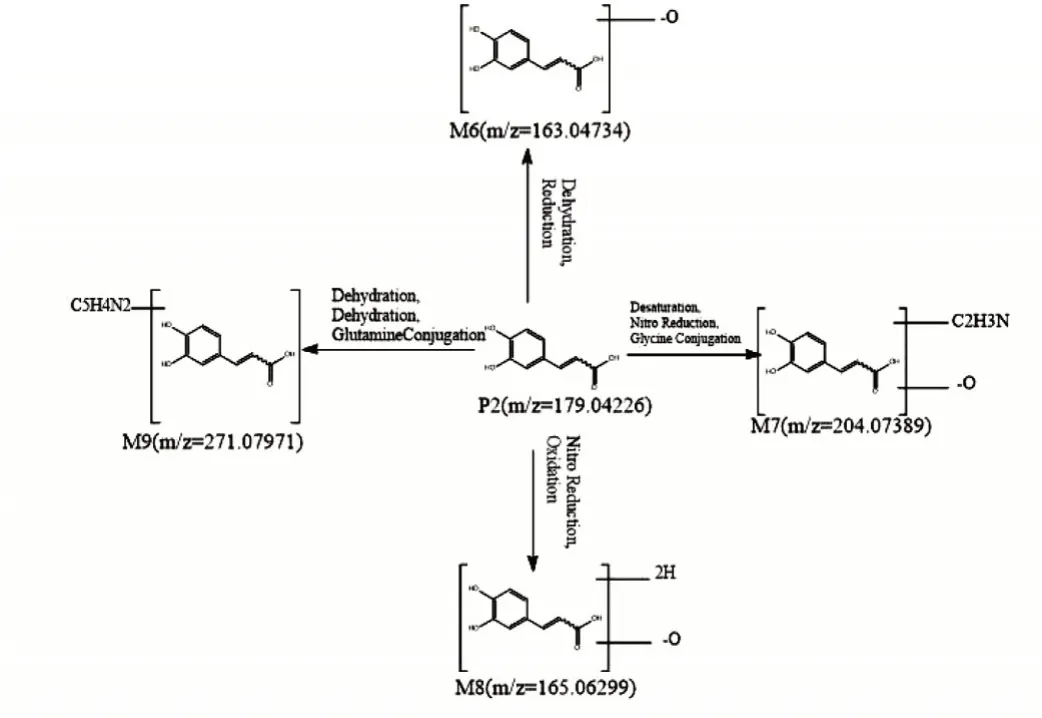
圖5 咖啡酸在大鼠體內代謝規律圖Fig.5 Metabolism of caffeic acid in rats
4 討論
中藥復方成分復雜,而且含量較低,因此進入體內的就更少,代謝產物就少之又少。Q-Exacive MS/MS技術不僅可以提供母離子及多級碎片子離子的精確分子量,且掃描速度快、靈敏度高,是作為體內成分鑒定的理想工具。軟件Compound discover可以輔助鑒定血中原型成分的所有Ⅰ相和Ⅱ相代謝產物[14]。兩者結合可以較全面地分析入血成分。生物樣本內源性成分對入血成分的檢測有很大的影響,因此需要對生物樣品進行前處理,減少內源性成分的干擾[15~16]。藥物在體內的代謝主要存在于肝組織中,代謝反應主要包括Ⅰ相代謝和Ⅱ相代謝。Ⅰ相代謝是機體經歷氧化、還原和水解反應,并將-OH、-COOH、-NH2、-SH和其他極性基團引入母體藥物。II相代謝是指藥物分子的極性基團和體內具有更大水溶性的內源性化合物如葡萄糖醛酸、硫酸、甘氨酸、谷胱甘肽等共價結合后,形成水溶性高、極性好的代謝物[17~18]。
從EDD的入血成分可以發現,吸收的成分主要集中在黃酮類和苯丙素類,另外還有個別環烯醚萜、倍半萜類等化合物,而且入血的母核也是苯丙素和黃酮類比較多。通過代謝規律分析發現,黃酮類主要的代謝反應有還原、水合、乙酰化、甲基化等I相代謝反應,有個別反應涉及到葡萄糖結合,氨基酸結合反應等II相反應。而苯丙素類化合物主要涉及脫水、羥基化、還原、硝基還原、氨基酸結合等II相代謝反應。總之,部分入血成分通過體內代謝反應增加其極性,便于經過尿液和膽汁排泄,EDD中大部分成分以原型成分進行吸收代謝。但是這些成分是否可以作為此方的藥效成分、是否具有這樣的代謝途徑,還需要進一步通過藥效實驗、代謝組學等相關方法進行驗證。
猜你喜歡
現代臨床醫學(2022年4期)2022-09-29 07:38:00
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
昆明醫科大學學報(2021年4期)2021-07-23 01:21:50
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
云南醫藥(2019年3期)2019-07-25 07:25:14
產品可靠性報告(2017年7期)2017-09-05 09:49:12
海南醫學(2016年8期)2016-06-08 05:43:00
汽車觀察(2016年3期)2016-02-28 13:16:26
醫學研究雜志(2015年9期)2015-07-01 17:28:15
