公用設施計算機化系統驗證研究與應用
2022-09-27 07:20:14錢貴珍
甘肅科技 2022年14期
芮 麗 ,王 哲,王 劍,錢貴珍
(1.蘭州生物制品研究所有限責任公司,甘肅 蘭州 730046;2.蘭州蘭生血液制品有限公司,甘肅 蘭州 730046)
隨著電子技術快速發展,公用設施計算機化系統廣泛應用于制藥企業的各輔助生產工藝環節,國內外對制藥行業《藥品生產質量管理規范》(GMP)相關的公用設施計算機化系統驗證提出了嚴格要求。計算機化系統的驗證是制藥行業在滿足患者安全、產品質量、數據完整性方面不可或缺的質量風險管理部分,單純的設備、工藝驗證不再滿足現有計算機化系統的合規管理,自2015年12月1日GMP附錄《計算機化系統》《驗證與確認》的實施[1],制藥企業所涉及的GMP相關計算機化系統的驗證越來越得到企業的重視,并成為國家藥監局重點審查對象[2]。文中以《良好自動化生產實踐指南》(GAMP 5)為指導,依據GMP附錄《計算機化系統》《確認與驗證》要求,結合實際工作經驗,對公用設施計算機化系統驗證進行深入研究及實例應用,以期為制藥企業公用設施計算機化系統驗證的實施提供參考。
1 計算機化系統
計算機化系統是由軟件、硬件(固件)、網絡組件和可控的功能以及人員和相關文件組成,如圖1所示。根據GAMP5,又將計算機化系統軟件分為基礎設施軟件、不可配置軟件、可配置軟件和定制應用軟件4種類別,硬件分為標準硬件組件和定制內置硬件組件[3]。
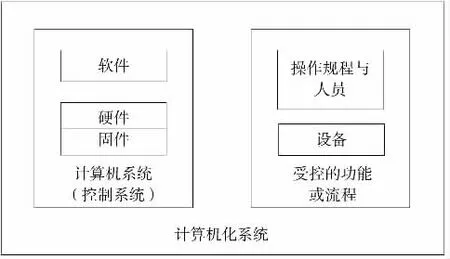
圖1 計算機化系統
2 公用設施計算機化系統
公用自動化系統通常是由多個單一設備構成,包括上位機、打印機、HMI、PLC、變頻器、閥門、傳感器等儀器儀表及現場設備。單個或多個自動化系統以及人員、流程、文件構成一個完整的計算機化系統。制藥企業公用設施計算機化系統一般包括:制水系統、空調自控系統、冷庫自控系統、生化培養罐系統、脈動真空滅菌器系統等,這些公用設施計算機化系統為制藥企業藥品生產提供良好的基礎輔助,并和產品質量相關聯,為了確保藥品生產過程符合《現行良好生產質量管理規范》(CGMP),與藥品生產相關的公用設施計算機化系統在整個生命周期內必須始終處于驗證狀態并穩定運行[4]。目前,制藥企業計算機化系統驗證主要參照國際制藥工程協會(ISPE)GAMP5。公用設施計算機化系統的軟件通常是為具體業務或工藝流程而定制的軟件產品,一般被歸類為GAMP類別4,通常采用由3個階段的規范與核實組成的驗證方法。涵蓋這3個階段所需要的文件數量要根據系統的復雜性與影響來確定。可配置產品(類別4)的驗證V模型,如圖2所示。

圖2 可配置產品的驗證V模型
3 公用設施計算機化系統驗證
本研究以公用系統冷庫計算機化系統為例進行驗證研究與應用分析。設計的冷庫計算機化系統包括:遠程監控系統、溫度控制器、調制解調器、制冷機組、冷庫等。用于儲存原材料、半成品、成品以及實驗用品等,系統通過溫度控制器控制制冷機組的運行啟停,上傳至遠程監控平臺實現對冷庫溫度的控制和監視,并生成報警記錄和事件記錄,關鍵溫度參數具有歷史趨勢查詢功能。制冷機組分別由冷風機、壓縮機、冷凝風機以及電磁閥等組成。圖3是典型的冷庫制冷系統電氣控制原理圖。
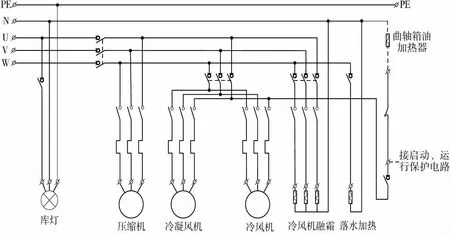
圖3 冷庫制冷系統電氣控制原理圖
3.1 驗證范圍確認
GMP附錄《計算機化系統》第四章第六條規定“計算機化系統驗證包括應用程序的驗證和基礎架構的確認,其范圍與程度應當基于科學的風險評估。風險評估應當充分考慮計算機化系統的適用范圍和用途。”[5]本研究通過系統差距分析和風險評估確定冷庫計算機化系統的驗證范圍與程度。
3.1.1 差距分析
GxP評估[6]冷庫計算機化系統為GMP關鍵,對于GMP關鍵系統進行計算機化系統驗證差距分析,首先通過系統的失效模式,分別從患者安全、產品質量、數據完整性3個方面的影響性來判定系統影響級別,如圖4所示。
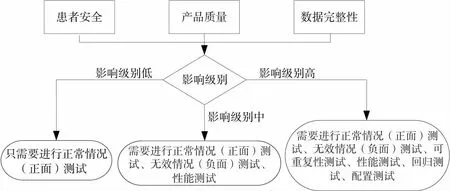
圖4 計算機化系統影響級別
通過系統的失效模式分析,判定冷庫計算機化系統影響因子為中,為直接影響系統,根據系統影響級別在差距分析及驗證措施中采取對應的測試活動。
差距分析采用現行冷庫計算機化系統目前狀態與中國GMP附錄《計算機化系統》、GAMP5的要求進行對比,通過法規要求提取問題進行分析,對提出的問題給出符合實際情況的最終結果以發現系統存在的潛在差距。差距分析的執行過程如圖5所示。
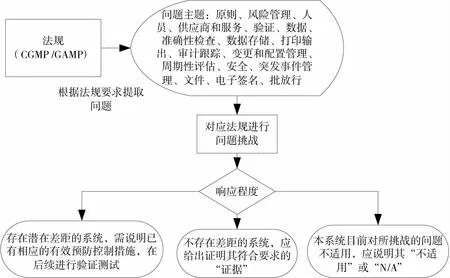
圖5 差距分析執行過程
基于上述分析所發現的差距,開展相關改進活動在后續的驗證及管理程序中加以處理和彌補,并在后續運行階段持續進行維護,從而使系統持續整體合規。
3.1.2 風險評估
對評估為直接影響的冷庫計算機化系統進行關鍵部件和功能評估,對判定為關鍵性的部件和功能進行風險評估以確定出所有的潛在危險及其對產品的影響。首先根據部件和功能對產品的影響來評估其GMP關鍵程度,部件和功能的GMP影響評估以產品的5個質量參數為基礎(功效、特性、安全、純度、質量),對于每一項會對產品質量產生影響的功能、所有提供該功能的設備、部件或儀表都歸類為關鍵和非關鍵兩種。部件和功能的關鍵性評估如表1所示。

表1 部件和功能的關鍵性評估
對歸類為關鍵的部件和功能應用失敗模式效果分析,對已經識別的或潛在的風險及問題進行分析,根據經驗和維修歷史數據進而確認每個風險的嚴重性(Severity)、發生的可能性(Probability of occurrence)及可檢測性(Detection),對風險進行深入的描述,并綜合上述因素確認一個風險的等級[7]。
根據風險等級與可接受標準的對應關系,做出是否對該風險采取措施的決定,對風險級別高的關鍵部件和功能進行風險控制,采取相應的糾正和預防措施后使風險處于可接受程度[8],在整個計算機化生命周期內使系統處于可控狀態。
3.2 設計功能驗證
計算機化系統設計功能(FS)驗證包括軟件設計功能(SDS)確認和硬件設計功能(HDS)確認,確認系統滿足用戶需求(URS)和CGMP的要求[9]。FS的編寫參照GAMP5-遵從GxP計算機化系統監管的風險管理方法中關于計算機化系統功能規范附錄D2。冷庫計算機化系統FS規范文件主要包含系統硬件、軟件結構,功能、數據、界面以及非功能屬性等。
3.2.1 硬件設計功能確認
冷庫計算機化系統硬件包括:操作員站上位機(主機、顯示器、鍵盤等)、現場溫度控制器、調制解調器、制冷機組、冷庫主體等主要部件。硬件功能確認需要對評估為計算機化系統的關鍵部件進行規格型號、供應商、技術參數的確認。確認冷庫計算機化系統滿足最初的硬件設計要求和用戶需求以及符合相關法規,并在實際運行過程中始終保持高效且符合現階段自動化系統技術要求。冷庫計算機化系統硬件結構如圖6所示。
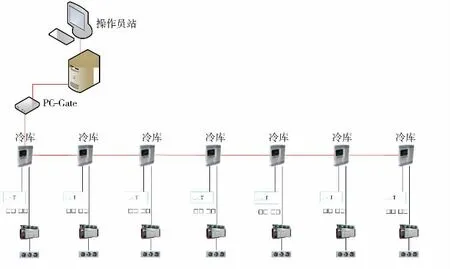
圖6 冷庫計算機化系統硬件結構
3.2.2 軟件設計功能確認
軟件設計功能確認通常包括:軟件結構、系統控制功能、數據處理功能、人機界面、系統訪問控制、系統安全,以及系統備份恢復等。對冷庫計算機化系統軟件功能進行確認使其滿足最初的軟件設計要求和用戶需求以及符合相關法規,并符合現階段系統智能化管理。
3.3 驗證方案確認
根據中國GMP附錄(2010修訂)《確認與驗證》《計算機化系統》的要求,通過驗證方案對運行階段的冷庫計算機化系統進行再次確認,以符合現行中國GMP(2010修訂)法規要求并保證系統正常、可靠地運行,以確保產品的存儲穩定性,使冷庫計算機化系統在運行階段始終保持驗證狀態。
驗證確認方案包含:安裝確認(IQ)、運行確認(OQ)和性能確認(PQ),具體見表2。

表2 驗證方案確認內容
3.4 驗證確認及報告階段
驗證確認階段主要是對驗證確認方案的實施,根據冷庫計算機化系統的功能規范要求,通過一系列的測試手段,確認系統硬件、軟件功能滿足設計要求、相關文件處于可控狀態、系統運行性能穩定可靠,滿足計算機化系統相關法規要求。
驗證確認完成后,對確認方案范圍、確認方案項目執行情況、項目完成情況以及偏差與變更情況進行總結得出結論,并依據中國GMP(2010修訂)第九十條規定“應當按照操作規程和校準計劃定期對生產和檢驗用衡器、量具、儀表、記錄和控制設備以及儀器進行校準和檢查,并保存相關記錄。校準的量程范圍應當涵蓋實際生產和檢驗的使用范圍。”,中國GMP(2010修訂)附錄《確認與驗證》第十六條規定“運行確認完成后,應當建立必要的操作、清潔、校準和預防性維護保養的操作規程,并對相關人員培訓。”以及中國GMP(2010修訂)附錄《計算機化系統》第二十條規定“企業應當建立應急方案,以便系統出現損壞時啟用。應急方案啟用的及時性應當與需要使用該方案的緊急程度相關。”等相關要求,對冷庫計算機化系統提出運行維護建議以滿足法規要求使冷庫計算機化系統始終處于驗證狀態,形成最終的確認報告。
4 結論
制藥企業公用設施計算機化系統運行多年,自GMP附錄《計算機化系統》2015年12月1日施行以來,制藥企業在這方面的驗證還不夠充分,本研究對運行階段的公用設施計算機化系統的驗證實施進行深入研究分析,使公用設施計算機化系統在滿足GMP法規要求的基礎上,對提升企業運營效益、降低管理成本做出突出的貢獻。
猜你喜歡
中華詩詞(2022年6期)2022-12-31 06:41:24
工業設計(2022年8期)2022-09-09 07:43:20
軍民兩用技術與產品(2021年10期)2021-03-16 06:05:30
北京測繪(2020年12期)2020-12-29 01:33:58
裝備制造技術(2019年12期)2019-12-25 03:06:46
中國洗滌用品工業(2019年4期)2019-05-11 09:27:34
家庭影院技術(2017年9期)2017-09-26 03:41:45
中國科技論壇(2017年7期)2017-07-25 08:49:53
媽媽寶寶(2017年2期)2017-02-21 01:21:24
國際漢語學報(2016年1期)2017-01-20 08:21:20
