NF-κB通路對口腔鱗癌細胞遷移和侵襲影響的初步研究
2022-09-28 13:46:44王爽邱林劉涵孫曉倩代曉華張軍劉浩
實用口腔醫學雜志 2022年5期
關鍵詞:水平
王爽 邱林 劉涵 孫曉倩 代曉華 張軍 劉浩
口腔癌是頭頸部最常見的惡性腫瘤,其中口腔鱗狀細胞癌(oral squamous cell carcinoma,OSCC)占比90%以上[1-2],5 年生存率約50%左右,與其侵襲性強及淋巴結轉移率高相關[3]。由于目前仍缺乏減少OSCC轉移的有效方法,故探究其相關轉移機制具有重要臨床意義。
據報道,慢性炎癥與OSCC發生和進展有關[4],而NF-κB通路作為炎癥相關癌癥調控中的經典通路,在包括OSCC在內的多數癌癥中被異常激活[5]。該通路通過調控下游細胞因子和趨化因子表達來促使腫瘤細胞發生上皮間充質轉化(epithelial-mesenchymal transition,EMT),隨之產生許多促炎因子,因此建立一個調節環,有利于維持EMT表型[5]。盡管NF-κB通路已經發現數十年,但在OSCC中的作用尚不清楚。
1 材料與方法
1.1 細胞培養
本實驗使用人舌鱗癌細胞系SCC-15(天津市口腔功能重建重點實驗室存儲),用含10%胎牛血清(Gibco,美國)和1%雙抗溶液(北京Solarbio)的高糖DMEM,在37 ℃和5%CO2濕潤培養箱中培養。
1.2 Transwell侵襲實驗
預冷無血清DMEM按1∶3稀釋Matrigel,取100 μL Matrigel膠均勻鋪在Transwell小室內,37 ℃孵育4 h至凝結成膠凍狀;設置20 μmol/L BAY11-7082預處理細胞1 h組、不預處理細胞組及PBS對照組,前兩組皆用1 μg/mL LPS刺激饑餓12 h以上的細胞24 h后收集細胞。用無血清DMEM將收集的細胞濃度調整為5×105/mL,上室加200 μL細胞懸液,下室加600 μL含20%血清的DMEM,培養24 h;棄小室中培養液,4%多聚甲醛固定15 min,PBS漂洗2 次,DAPI室溫避光染色5 min,倒置熒光顯微鏡下觀察,每個小室隨機拍攝5 個視野并計數。
1.3 裂痕愈合實驗
細胞分組及處理同上,收集細胞后按7×104個細胞/孔接種到傷口愈合檢測皿(ibidi, 德國)中,待細胞長滿后拔掉皿中自帶插件形成細胞鋪展面裂痕,換成無血清培養基,初始傷口寬度為500 μm(0 h),細胞培養24 h,棄培養基,PBS漂洗后用倒置熒光顯微鏡采集裂痕寬度圖像,Image J軟件測量傷口閉合面積。
1.4 蛋白質免疫印跡(Western blot)
饑餓細胞12 h以上,用不同濃度的LPS處理細胞指定時間后收集細胞樣品,等體積PBS作為對照。在4 ℃下提取細胞蛋白,BCA蛋白檢測試劑盒測定蛋白濃度。用SDS-PAGE預制膠分離蛋白質,電轉至PVDF膜,5%脫脂奶室溫封閉1 h;用IκBα、p65、p-p65、ZEB2(1∶500,Santa Cruz,California,USA)、Tubulin、E-cadherin、Vimentin(1∶1 000,上海,Beyotime)4 ℃搖床孵育過夜;1×TBS漂10 min共3 次,HRP標記的山羊抗兔/小鼠IgG室溫搖床(1∶1 000,Beyotime)孵育1 h,1×TBS漂10min共3次。化學發光試劑顯色后用Image Lab自動凝膠分析軟件對條帶進行定量。
1.5 實時熒光定量PCR(real-time PCR)
饑餓細胞12 h以上,用1 μg/mL LPS處理不同時間段(0、0.5、1、2、3、4、5、6、8、16、24 h)后收集細胞,等體積PBS作為對照。提取細胞總RNA反轉錄后經Light Cycler/Light Cycler 480系統(羅氏,瑞士)進行RT-PCR擴增。GAPDH為內參,2-ΔΔCt法計算mRNA表達相對倍數變化。引物(中國生工生物技術)見表1。

表1 引物序列
1.6 免疫熒光染色
饑餓細胞12 h以上,用1 μg/mL LPS刺激8 h后收集細胞,等體積PBS作為對照。按8×104個細胞/孔接種到共聚焦小皿中過夜后4%多聚甲醛固定15 min,PBS漂洗5 min共3 次;室溫封閉15 min;4 ℃中與p65抗體孵育過夜后PBS漂洗15 min,室溫下Cy3(紅色)偶聯熒光二抗(1∶200,Beyotime)避光染色1 h,PBS漂洗;DAPI避光染色5 min,PBS去浮色,倒置熒光顯微鏡對染色結果成像。
1.7 統計學分析
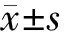
2 結 果
2.1 LPS激活SCC-15細胞NF-κB通路
SCC-15細胞經1 μg/mL LPS處理0、0.5、1、2、4、8、16和24 h后,IκBα 蛋白表達量在4 h到8 h顯著降低(圖1A);mRNA表達量在4~8 h顯著升高(P<0.05),5 h時達到峰值,之后逐漸下降(圖1B)。結果提示,IκBα作為NF-κB通路的負反饋調節因子,在LPS刺激SCC-15細胞4 h后蛋白表達水平降低,從而激活NF-κB通路;免疫熒光結果顯示,對照組中p65核中染色弱(紅色熒光),而處理組p65核中染色強(圖1C),LPS誘導 p65從細胞質向核轉運。上述結果表明LPS成功誘導NF-κB通路活化。

圖1 LPS激活SCC-15細胞的NF-κB通路
2.2 NF-κB通路調控SCC-15細胞遷移和侵襲
傷口愈合實驗顯示,相比對照組,SCC-15細胞遷移能力提高了1.785 倍(P<0.05,圖2A);Transwell侵襲實驗中,相比對照組,SCC-15細胞侵襲能力提高56.4%±7.2%(P<0.05,圖2B)。但NF-κB通路抑制劑BAY11-7082處理細胞后,相比LPS處理組,BAY11-7082組顯著降低SCC-15細胞的相對遷移面積(P<0.05,圖2C)以及穿膜細胞數量,平均每個視野中穿膜細胞數不足LPS組的1/4(P<0.05,圖2D)。結果表明,NF-κB通路活化促進SCC-15細胞遷移和侵襲。

圖2 NF-κB通路調控SCC-15細胞遷移和侵襲
2.3 LPS上調SCC-15細胞中p65磷酸化蛋白及促炎因子
p65 mRNA表達水平在經LPS刺激0、0.5、1、2、3、4、5、6、8、16和24 h后均無明顯變化(P>0.05, 圖3A);而p65磷酸化蛋白表達水平分別呈LPS刺激時間和濃度依賴性,24 h達峰值,較對照組上調3.39 倍,0.1 μg/mL LPS 處理下即可顯著上調其磷酸化蛋白,1 μg/mL和10 μg/mL LPS處理時上調更為顯著(圖3B~3C)。這表明,NF-κB通路活性可能與SCC-15細胞中p65磷酸化蛋白水平相關。
通過RT-PCR檢測相同刺激條件下IL-6和COX-2 mRNA表達,發現相比對照組,處理組顯著性上調IL-6和COX-2 mRNA表達水平(P<0.05)。其中,IL-6在LPS處理0.5 h后達到峰值(3.71 倍),隨后上調量逐漸減少,5 h后恢復至對照組水平(圖3D);COX-2在LPS處理0.5 h后顯著上調,至5 h達到峰值(10.2 倍),之后緩慢降低,24 h時恢復至對照組水平(圖3E)。上述結果提示LPS促進SCC-15細胞中促炎因子表達。

圖3 LPS上調SCC-15細胞中p65磷酸化蛋白及促炎因子
2.4 LPS促進SCC-15細胞EMT
濃度為1 μg/mL以上的LPS處理SCC-15細胞24 h后,相比對照組,處理組上皮標記物E-cadherin表達顯著降低,而間充質標記物Vimentin和ZEB2表達水平顯著升高(P<0.05,圖4),表明LPS促進SCC-15細胞發生EMT。

圖4 LPS促進SCC-15細胞EMT
3 討 論
目前已知NF-κB家族由5 個成員組成:RELA(p65)、RELB、c-REL、p50/p105(NF-κB1)和p52/p100(NF-κB2),它們可以形成二聚物復合物。這些復合物被其抑制蛋白(IκBs)限制在胞質中,可被各種刺激(如細胞因子、細菌感染等)激活易位至胞核,進而激活下游促炎因子(如IL-6和COX-2等)[6]。本實驗結果顯示,經LPS誘導后,p65在胞核中明顯表達,且SCC-15細胞中IκBα mRNA在4~6 h再合成,蛋白在4~8 h顯著降解,這表明IκBα mRNA可翻譯成活性的IκBα蛋白,蛋白降解和再合成的量相當,而在8 h后IκBα蛋白維持在對照組水平;同時p65(Ser311)磷酸化蛋白水平呈持續性上調,這說明NF-κB通路持續響應LPS刺激而處于轉錄激活狀態。
NF-κB是參與炎癥相關癌癥調節的中心信號通路,該通路的異常活化能夠加速癌癥發展[7]。IL-6具有腫瘤生長促進作用,在OSCC中有明顯的高表達且與p65的表達呈正相關[8]。相比于正常口腔黏膜,OSCC中COX-2高表達并與腫瘤局部復發、淋巴結轉移有關;同時COX-2可上調細胞間粘附分子-1(ICAM-1)和血管內皮生長因子C(VEGFC),進而促進OSCC轉移[9-10]。在本研究中,LPS激活組中SCC-15細胞遷移和侵襲性較對照組明顯增強,而用BAY11-7082抑制NF-κB通路后,細胞遷移和侵襲性明顯減弱,這表明NF-κB通路可能調節OSCC細胞遷移和侵襲。
惡性腫瘤轉移是一個多步驟過程[11],其中通過EMT過程能使細胞間粘附喪失,同時能促進遷移和侵襲所需的形態和行為轉化[12]。炎癥中的可溶性因子可以促進癌細胞獲得EMT相關的運動性、侵襲性和降解細胞外基質等特征[13]。在本研究中,經LPS刺激,發現維持正常上皮細胞間粘附的E-cadherin表達顯著下調,同時Vimentin和ZEB2表達顯著上調,由于Vimentin的高表達與OSCC轉移潛力增加相關[14],且NF-κB可通過刺激轉錄因子ZEB2轉錄來抑制E-cadherin表達[15],因此使得SCC-15細胞具有更強的遷移、侵襲潛能,表明激活的NF-κB通路可能通過調節EMT過程促進OSCC遷移和侵襲。
總之,本文通過體外研究初步證實炎癥通路NF-κB可以調節EMT過程且促進OSCC遷移和侵襲,為闡述NF-κB通路在OSCC中的作用提供部分實驗證據。
猜你喜歡
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
火花(2019年12期)2019-12-26 01:00:28
人大建設(2019年6期)2019-10-08 08:55:48
人大建設(2019年12期)2019-05-21 02:55:32
雜文月刊(2018年21期)2019-01-05 05:55:28
人大建設(2017年6期)2017-09-26 11:50:44
學苑創造·A版(2015年11期)2016-01-14 09:03:27
俄羅斯問題研究(2012年1期)2012-03-25 09:54:45
中國火炬(2010年12期)2010-07-25 13:26:22
中國火炬(2010年8期)2010-07-25 11:34:30
