固腎定喘丸多指標成分一測多評法的建立及質量評價Δ
2022-09-30 14:26:12李志平高雨秋王加良侯甲福牡丹江醫學院附屬紅旗醫院藥學部黑龍江牡丹江570牡丹江醫學院藥學院黑龍江牡丹江570
中國藥房 2022年18期
張 穎,李志平,高雨秋,王加良,侯甲福(.牡丹江醫學院附屬紅旗醫院藥學部,黑龍江 牡丹江 570;.牡丹江醫學院藥學院,黑龍江 牡丹江 570)
固腎定喘丸由肉桂、熟地黃、車前子、鹽補骨脂、附片、鹽益智仁等13味中藥飲片加工而成,方中鹽補骨脂溫腎助陽、納氣平喘,為君藥;附片和肉桂補腎陽、固腎定喘,鹽益智仁和金櫻子肉溫,補脾腎,合為臣藥;熟地黃、山藥、茯苓、牡丹皮和澤瀉滋補腎陰、滲濕熱,車前子和牛膝利水滲濕、補肝腎,合為佐藥;砂仁化濕開胃、溫脾止瀉,為使藥。固腎定喘丸可溫腎納氣、健脾化痰,臨床主要用于治療肺脾氣虛、腎不納氣所致的咳嗽和氣喘,亦可用于慢性支氣管炎、肺氣腫、支氣管哮喘而引起的咳嗽和氣喘。固腎定喘丸收載于2020年版《中國藥典》(一部)[1],其質量標準及文獻報道[2]僅對君藥鹽補骨脂中補骨脂素進行了定量分析。對于所含化學成分復雜、臨床作用機制不清晰的中藥復方制劑來說,檢測指標過于單一,對體現中藥制劑的整體質量、保證其產品質量一致性存在一定的局限。
近年來,一測多評(quantitative analysis of multicomponents by single marker,QAMS)法的定量分析越來越多地應用于中成藥復方制劑質量評價中[3]。化學模式識別分析可通過統計學或數學方法,挖掘復雜數據間存在的內在函數關系,現已廣泛應用于中藥多維信息的綜合分析中[4]。本實驗參考中藥質量標志物確認依據,選取純度較高、質量穩定、價廉易得的補骨脂素為內參物,采用QAMS法對固腎定喘丸中君藥鹽補骨脂特征成分補骨脂素、異補骨脂素、新補骨脂異黃酮和補骨脂二氫黃酮,臣藥肉桂藥效成分肉桂酸和桂皮醛,佐藥熟地黃和車前子主要成分大車前苷、毛蕊花糖苷、異毛蕊花糖苷和木通苯乙醇苷B共10個成分的含量進行檢測,建立固腎定喘丸多指標成分一測多評法,同時利用化學模式識別分析方法對不同批次樣品的檢測結果進行評價,以期完善并提高固腎定喘丸的質量控制手段,確保產品質量穩定和臨床療效的一致性。
1 材料
1.1 主要儀器
本研究所用主要儀器有1200型高效液相色譜(HPLC)儀(美國Agilent公司)、2695型HPLC儀(美國Waters公司)、MS105DU型電子天平(瑞士Mettler Toledo公司)、SB-5200DTDN型超聲波清洗機(寧波新芝生物科技有限公司)等。
1.2 主要藥品與試劑
固腎定喘丸(水蜜丸,國藥準字Z44020906)購自廣州白云山敬修堂藥業股份有限公司,批號分別為L02008、L03011、L03016、L04018、L06035、L06041、L08050、M04001、M12021、M12050、Y04007、Y10010、T12001、F08027和H10028,編號依次為S1~S15。肉桂酸對照品(批號110786-201604,純度98.8%)、桂皮醛對照品(批號110710-202022,純度99.5%)、大車前苷對照品(批號111914-202105,純度96.0%)、毛蕊花糖苷對照品(批號111530-201914,純度95.2%)、木通苯乙醇苷B對照品(批號111910-201604,純度98.2%)、補骨脂素對照品(批號110739-201918,純度99.6%)、異補骨脂素對照品(批號110738-202016,純度99.4%)、新補骨脂異黃酮對照品(批號520052-201401,純度99.6%)和補骨脂二氫黃酮對照品(批號520053-201401,純度99.4%)均購自中國食品藥品檢定研究院;異毛蕊花糖苷對照品(批號PRF9010243,純度97.0%)購自成都普瑞法科技開發有限公司;乙腈、甲醇均為色譜純,其余試劑均為分析純。
2 方法與結果
2.1 色譜條件
以Agilent SB-C18(250 mm×4.6 mm,5 μm)為色譜柱,以乙腈(A)-0.2%冰醋酸溶液(B)為流動相進行梯度洗脫(0~11 min,25%A;11~20 min,25%A→40%A;20~35 min,40%A→50%A;35~59 min,50%A→75%A;59~70 min,75%A→25%A);柱溫為30℃;流速為1.0 mL/min;檢測波長分別為290 nm(0~20 min,檢測肉桂酸和桂皮醛)[5-6]、330 nm(20~35 min,檢測大車前苷、毛蕊花糖苷、異毛蕊花糖苷和木通苯乙醇苷B)[7-9]和246 nm(35~70 min,檢測補骨脂素、異補骨脂素、新補骨脂異黃酮和補骨脂二氫黃酮)[10-11];進樣量為10μL。
2.2 溶液的制備
2.2.1 混合對照品溶液 精密稱取肉桂酸、桂皮醛、大車前苷、毛蕊花糖苷、異毛蕊花糖苷、木通苯乙醇苷B、補骨脂素、異補骨脂素、新補骨脂異黃酮和補骨脂二氫黃酮對照品適量,用甲醇溶解制成質量濃度分別為0.198、0.472、0.296、2.130、0.414、0.136、1.408、0.960、0.572、0.538 mg/mL的混合對照品貯備液。將混合對照品貯備液用甲醇稀釋20倍制得混合對照品溶液(上述成分質量濃度分別為9.9、23.6、14.8、106.5、20.7、6.8、70.4、48.0、28.6、26.9 μg/mL)。
2.2.2 供試品溶液 取固腎定喘丸適量,研細,取約3 g,精密稱定,置具塞錐形瓶中,精密加入甲醇25 mL,稱定質量后,水浴回流提取60 min,放冷,用甲醇補足減失質量,搖勻,經0.45 μm微孔濾膜過濾,取續濾液,即得。
2.2.3 陰性供試品溶液 取按固腎定喘丸標準處方和制法制備的缺肉桂、缺鹽補骨脂、缺熟地黃和車前子的3種陰性供試品各適量,按“2.2.2”項下方法制得相應陰性供試品溶液。
2.3 方法學考察
2.3.1 專屬性考察 取混合對照品溶液、供試品溶液和陰性供試品溶液,按“2.1”項下色譜條件進樣測定,記錄色譜圖(圖1)。結果顯示,供試品溶液中各待測成分與相鄰色譜峰分離良好(分離度均不小于1.5),理論板數按各成分色譜峰計均不小于5 500,陰性供試品對測定無干擾。
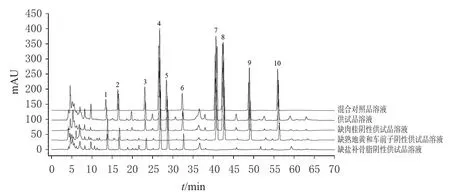
圖1 固腎定喘丸定量分析的HPLC圖
2.3.2 線性關系考察 精密吸取混合對照品貯備液0.1、0.2、0.5、1.0、2.0、5.0 mL,置于不同的20 mL量瓶中,用甲醇稀釋制得6個系列質量濃度的混合對照品溶液,按“2.1”項下色譜條件進樣測定,記錄峰面積。以各待測成分質量濃度為橫坐標(X)、峰面積為縱坐標(Y)進行線性回歸,結果見表1。

表1 固腎定喘丸中10個待測成分的回歸方程和線性范圍
2.3.3 精密度試驗 取供試品溶液(編號S1),按“2.1”項下色譜條件連續進樣6次,記錄峰面積。結果顯示,肉桂酸等10個待測成分峰面積的RSD均小于2.00%(n=6),表明方法精密度良好。
2.3.4 重復性試驗 精密稱取同一批固腎定喘丸樣品(編號S1)6份,分別按“2.2.2”項下方法制備供試品溶液,按“2.1”項下色譜條件進樣測定,記錄峰面積并用外標法計算含量。結果顯示,肉桂酸等10個待測成分含量的RSD均小于2.00%(n=6),表明方法重復性良好。
2.3.5 穩定性試驗 取供試品溶液(編號S1),分別在室溫下放置0、2、4、6、12、24 h時按“2.1”項下色譜條件進樣測定,記錄峰面積。結果顯示,肉桂酸等10個待測成分峰面積的RSD均小于2.00%(n=6),表明供試品溶液在室溫下放置24 h內穩定性良好。
2.3.6 加樣回收率試驗 取已知待測成分含量的固腎定喘丸樣品(編號S1)適量,研細,分別取9份,每份精密稱定1.5 g,均分成3組,每組分別精密加入混合對照品溶液(肉桂酸、桂皮醛、大車前苷、毛蕊花糖苷、異毛蕊花糖苷、木通苯乙醇苷B、補骨脂素、異補骨脂素、新補骨脂異黃酮和補骨脂二氫黃酮的質量濃度分別為0.107、0.351、0.232、1.427、0.278、0.072、0.931、0.709、0.435 和0.379 mg/mL)0.8、1.0、1.2 mL,按“2.2.2”項下方法制備供試品溶液,再按“2.1”項下色譜條件進樣測定,記錄峰面積并計算加樣回收率。結果顯示,肉桂酸等上述10個待測成分的平均加樣回收率為96.98%~100.09%,RSD均小于2.00%(n=9),表明方法準確度良好。
2.4 QAMS法的建立
2.4.1 相對校正因子的計算 用對照品質量濃度與峰面積之比計算各待測成分的相對校正因子(relative correction factor,RCF),即RCF=(Wk×As)/(Ws×Ak)(式中W和A分別為質量濃度和峰面積,下標k和s為其他待測成分和內參物)。按“2.1”項下色譜條件進樣測定“2.3.2”項下6個系列質量濃度的混合對照品溶液,以補骨脂素為內參物,計算得肉桂酸、桂皮醛、大車前苷、毛蕊花糖苷、異毛蕊花糖苷、木通苯乙醇苷B、異補骨脂素、新補骨脂異黃酮和補骨脂二氫黃酮的RCF分別為2.818 7、1.567 8、2.202 3、1.227 0、1.744 4、4.466 7、0.970 0、1.084 8和1.257 7,RSD均小于2.00%(n=6)。
2.4.2 RCF耐用性試驗 精密吸取“2.2.1”項下混合對照品溶液,按“2.1”項下色譜條件進樣測定,記錄峰面積,分別考察不同HPLC儀(Waters 2695型、Agilent 1200型,下同)、不同色譜柱[Agilent SB-C18、Phenomenex SuperLu C18、Welch Ultimate XB C18,規格均為(250 mm×4.6 mm,5 μm),下同]、不同流速(0.8、1.0、1.2 mL/min)及不同柱溫(25、30、35℃)對所建立的RCF的影響。結果顯示,不同儀器及不同色譜柱下肉桂酸等上述9個待測成分的平均RCF分別為2.816 4、1.571 5、2.204 5、1.222 4、1.743 5、4.463 6、0.968 1、1.079 0和1.254 0,不同流速下的平均RCF分別為2.8147、1.5606、2.2083、1.2278、1.7462、4.468 9、0.971 1、1.081 7和1.259 2,不同柱溫下的平均RCF 分別為 2.813 1、1.564 3、2.218 0、1.225 6、1.742 6、4.466 0、0.963 8、1.082 9和1.255 0,RSD均小于2.00%(n=9),表明RCF耐用性良好。
2.4.3 色譜峰定位 采用相對保留時間值(relative retention time,RRT)法對待測成分色譜峰進行定位,精密吸取“2.2.1”項下混合對照品溶液,按“2.1”項下色譜條件進樣測定,以補骨脂素為內參物,考察不同儀器和不同色譜柱對肉桂酸等上述9個待測成分的RRT的影響。結果顯示,9個待測成分的RRT分別為0.332 0、0.405 2、0.563 9、0.653 8、0.699 9、0.794 8、1.041 9、1.203 2 和1.380 6,RSD均小于2.00%(n=6)。
2.5 含量測定
取15批固腎定喘丸樣品(編號S1~S15),分別按“2.2.2”項下方法制備供試品溶液,按“2.1”項下色譜條件進樣測定,記錄峰面積,分別采用外標法和QAMS法計算樣品中上述10個待測成分的含量(表2)。采用SPSS 26.0軟件進行t檢驗,比較2種方法檢測結果的差異。結果顯示,2種方法的檢測結果差異無統計學意義(P>0.05)。

表2 15批固腎定喘丸中10個待測成分含量測定結果(mg/g,n=3)
2.6 固腎定喘丸化學模式識別分析方法的建立
2.6.1 聚類分析 將“2.5”項下15批固腎定喘丸中10個待測成分QAMS法含量數據導入SPSS 26.0軟件進行聚類分析(cluster analysis,CA)。結果顯示,當歐氏距離為15時,15批樣品聚為3類,S1~S7為第Ⅰ類,S8~S10為第Ⅱ類,S11~S15為第Ⅲ類。
2.6.2 主成分分析 將“2.5”項下15批固腎定喘丸中10個待測成分QAMS法含量數據導入SPSS 26.0軟件進行主成分分析(principal component analysis,PCA)。結果顯示,前2個主成分特征值分別為6.391和2.654,對主成分的方差貢獻率分別為63.905%和26.541%,累計方差貢獻率為90.446%(大于85%),表明選取前2個主成分即可代表固腎定喘丸90.446%的信息量。第一主成分的信息來自肉桂酸、大車前苷、毛蕊花糖苷、異毛蕊花糖苷、木通苯乙醇苷B、新補骨脂異黃酮和補骨脂二氫黃酮的綜合,第二主成分的信息來自桂皮醛、補骨脂素和異補骨脂素的綜合。同時應用SIMCA 14.1軟件建立PCA模型,得15批固腎定喘丸樣品的PCA得分圖(圖2)。從圖2可以看出,15批樣品聚為3類,與CA結果一致。
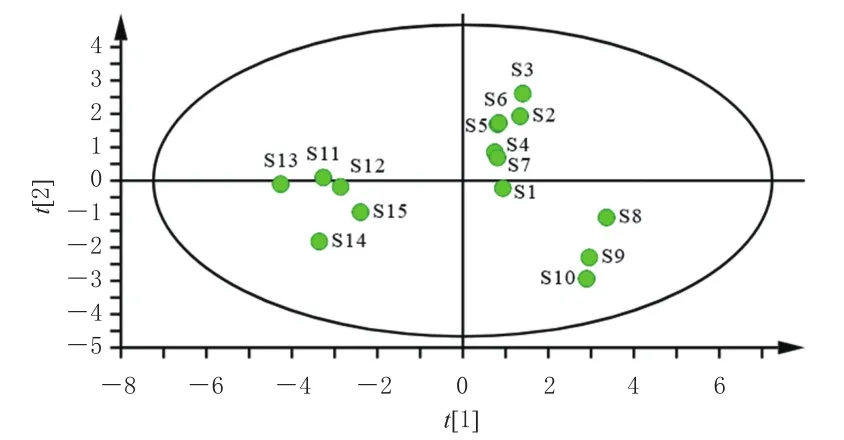
圖2 15批固腎定喘丸樣品的PCA得分圖
2.6.3 偏最小二乘法-判別分析 將“2.5”項下15批固腎定喘丸中10個待測成分QAMS法含量數據導入SIMCA 14.1軟件,運行偏最小二乘法-判別分析(partial least squares discrimination analysis,PLS-DA)程序,得圖3。由圖3可知,模型的累積解釋能力參數(R2X、R2Y)分別為0.917和0.851,預測能力參數(Q2)為0.789,均大于0.5,說明所建立的模型穩定可靠、預測能力強[12],可用于區分不同批次的固腎定喘丸。
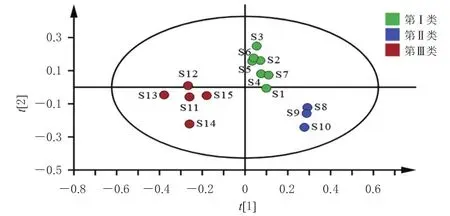
圖3 15批固腎定喘丸樣品的PLS-DA模型得分圖
對建立的PLS-DA模型進行200次置換檢驗(圖4),結果顯示,R2擬合直線Y軸截距為0.076 7,小于0.3,表明所建立的模型結果可靠;Q2擬合直線Y軸截距為-0.296,小于0.05,表明所建立的模型不存在過度擬合,可有效判別分析15批固腎定喘丸的質量差異。根據變量重要性投影(variable importance in projection,VIP)值篩選影響固腎定喘丸質量的標志性成分,結果顯示,補骨脂素、毛蕊花糖苷、桂皮醛和異補骨脂素是影響該藥質量的標志性成分(VIP值均大于1.0)。
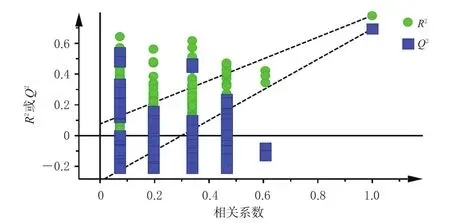
圖4 15批固腎定喘丸樣品的PLS-DA模型置換檢驗圖
3 討論
本實驗在制備供試品溶液時,考察了不同提取方法(超聲提取和水浴回流提取)、不同提取溶劑(水、甲醇和乙醇)、不同提取時間(30、60、90 min)對固腎定喘丸樣品中10個待測成分提取率的影響及雜質干擾情況。結果顯示,水浴回流提取效率較高;溶劑為甲醇時,10個待測成分的響應值較大;提取時間為60 min時,提取效率最高,同時雜質最少。綜合以上條件,選取甲醇水浴回流提取60 min為最佳提取方式。
本實驗在篩選流動相時,首先以甲醇-水、乙腈-水為流動相,發現以甲醇-水為流動相時,檢測用時較長,且數個色譜峰分離度達不到要求;以乙腈-水為流動相時,毛蕊花糖苷、補骨脂素和補骨脂二氫黃酮色譜峰出現拖尾現象,考慮用酸類溶液加以改善。通過對比乙腈-0.1%磷酸溶液[13]、乙腈-0.1%甲酸溶液[14]、乙腈-0.2%冰醋酸溶液[15]為流動相時10個待測成分的分離效果,結果顯示,以乙腈-0.2%冰醋酸溶液為流動相時,10個待測成分色譜峰均可達到基線分離且峰形較好。因此,選擇乙腈-0.2%冰醋酸溶液為流動相對固腎定喘丸中10個待測成分同時進行含量測定。
外標法和QAMS法所得10個待測成分含量測定結果差異無統計學意義(P>0.05),表明本研究建立的QAMS法較為合理。從CA、PCA和PLS-DA結果可以看出,15批固腎定喘丸樣品可聚為3類,君藥所含成分補骨脂素和異補骨脂素,臣藥所含成分桂皮醛及佐藥所含成分毛蕊花糖苷是影響固腎定喘丸產品質量的潛在標志性成分。綜上所述,本研究所建立的QAMS法多指標成分定量控制及化學模式識別分析可用于固腎定喘丸的質量評價。
