雙醋瑞因膠囊在健康受試者中的餐后生物等效性研究Δ
2022-09-30 14:26:18徐鳳華黃明南京醫(yī)科大學(xué)附屬蘇州科技城醫(yī)院藥學(xué)部江蘇蘇州215153蘇州大學(xué)附屬第二醫(yī)院臨床藥理實(shí)驗(yàn)室江蘇蘇州215151
中國藥房 2022年18期
徐鳳華,黃明(1.南京醫(yī)科大學(xué)附屬蘇州科技城醫(yī)院藥學(xué)部,江蘇 蘇州 215153;2.蘇州大學(xué)附屬第二醫(yī)院臨床藥理實(shí)驗(yàn)室,江蘇 蘇州 215151)
雙醋瑞因膠囊原研制劑(商品名安必丁?)由阿根廷TRB Pharma S.A.公司研發(fā),是骨關(guān)節(jié)炎白細(xì)胞介素1(interleukin-1,IL-1)的主要抑制劑,用于髖、膝關(guān)節(jié)等骨關(guān)節(jié)炎的臨床治療[1]。有研究指出,該藥對(duì)骨關(guān)節(jié)炎患者的關(guān)節(jié)功能具有顯著的保護(hù)作用,可通過減輕疼痛、延緩病程來提高患者的生活質(zhì)量,且較非甾體抗炎藥有更強(qiáng)的后續(xù)效應(yīng)和更高的安全性[1-2]。
藥動(dòng)學(xué)研究和仿制藥一致性評(píng)價(jià)均需要測(cè)定藥物的藥動(dòng)學(xué)參數(shù),故準(zhǔn)確檢測(cè)生物樣品中的藥物濃度顯得非常重要。按照我國藥品注冊(cè)管理要求,仿制藥注冊(cè)需在健康人體中進(jìn)行生物等效性研究[3]。目前,雙醋瑞因膠囊仿制藥在中國健康受試者空腹?fàn)顟B(tài)下的藥動(dòng)學(xué)數(shù)據(jù)已有報(bào)道[4]。參考上述研究,結(jié)合原研制劑說明書推薦(雙醋瑞因宜餐后服用),本研究擬采用甲醇沉淀蛋白預(yù)處理血漿樣品,使用液相色譜-串聯(lián)質(zhì)譜(HPLC-MS/MS)法測(cè)定人血漿中雙醋瑞因活性代謝產(chǎn)物大黃酸的濃度;同時(shí),擬初步評(píng)價(jià)雙醋瑞因膠囊原研制劑(參比制劑)與國產(chǎn)仿制藥(受試制劑)在中國健康受試者體內(nèi)的餐后生物等效性,以期為國產(chǎn)雙醋瑞因膠囊的上市及臨床應(yīng)用提供參考依據(jù)。
1 材料
1.1 主要儀器
本研究所用主要儀器包括1200型高效液相色譜儀(美國Agilent公司),Turboionspray?型電噴霧離子源、API4000型三重四極桿串聯(lián)質(zhì)譜儀、Analyst 1.6數(shù)據(jù)采集軟件(美國AB Sciex公司),XS 105DU型分析天平(瑞士Mettler Toledo公司),Milli-Q Academic型純水機(jī)(美國Millipore公司)等。
1.2 主要藥品與試劑
本研究所用受試制劑雙醋瑞因膠囊(批號(hào)T93-013,規(guī)格50 mg)由江蘇某制藥有限公司提供;參比制劑雙醋瑞因膠囊(商品名安必丁?,國藥準(zhǔn)字HJ20150130,規(guī)格50 mg)由阿根廷TRB Pharma S.A.公司生產(chǎn)并由昆明積大制藥股份有限公司分裝。大黃酸對(duì)照品(批號(hào)110757-200206,純度100.0%)、大黃素對(duì)照品(內(nèi)標(biāo),批號(hào)110756-200110,純度100.0%)均購自中國食品藥品檢定研究院;甲醇和甲酸均為色譜純,氫氧化鈉和醋酸銨均為分析純,水為超純水。
1.3 空白血漿
空白血漿來自本研究所納入的健康受試者,于其給藥前采集、處理所得。
2 方法與結(jié)果
2.1 色譜與質(zhì)譜條件
色譜柱為Agilent ZORBAX Eclipse plus C8(4.6 mm×100 mm,3.5 μm),保護(hù)柱為Phenomenex Security GuardTMC18(4 mm×3.0 mm);流動(dòng)相為甲醇(A)-含0.4%甲酸的5 mmoL/L醋酸銨溶液(B),梯度洗脫(0~4.1 min,75%A;4.1~4.2 min,75%A→95%A;4.2~5.2 min,95%A;5.2~5.3 min,95%A→75%A;5.3~8.0 min,75%A);流速為1 000 μL/min;柱溫為30 ℃;進(jìn)樣量為10 μL;分析時(shí)間為8.0 min。
離子源為電噴霧離子源(electrospray ionization,ESI),采用多反應(yīng)監(jiān)測(cè)(multi-reaction monitoring,MRM)模式進(jìn)行負(fù)離子掃描;霧化氣壓力為344 738 Pa,加熱輔助氣壓力為344 738 Pa;離子源溫度為550℃;電噴霧電壓為-3 500 V;大黃酸和內(nèi)標(biāo)的定量離子對(duì)分別為m/z283.0→238.8、m/z268.9→224.8,去簇電壓分別為-50、-125 V,射入電壓分別為-3、-10 V,碰撞電壓分別為-35、-50 V,碰撞室射出電壓均為-5 V。
2.2 大黃酸和內(nèi)標(biāo)相關(guān)溶液的配制
2.2.1 大黃酸貯備溶液及工作溶液 稱取大黃酸對(duì)照品10.00 mg,轉(zhuǎn)移至10 mL量瓶中,加入0.5 mol/L氫氧化鈉溶液20 μL,再用甲醇定容,混勻,得質(zhì)量濃度為1.0 mg/mL的大黃酸標(biāo)準(zhǔn)曲線貯備溶液。取上述貯備溶液適量,用甲醇稀釋,得質(zhì)量濃度分別為200.0、100.0、40.00、20.00、10.00、4.000、2.000、0.600 0 μg/mL 的大黃酸標(biāo)準(zhǔn)曲線工作溶液,160.0、16.00、1.600 μg/mL的大黃酸質(zhì)控工作溶液和0.600 0 μg/mL的大黃酸定量下限工作溶液。
2.2.2 內(nèi)標(biāo)貯備溶液及工作溶液 稱取內(nèi)標(biāo)對(duì)照品10.00 mg,轉(zhuǎn)移至10 mL量瓶中,用甲醇定容,混勻,得質(zhì)量濃度為1.0 mg/mL的內(nèi)標(biāo)貯備溶液。取上述貯備溶液適量,用甲醇稀釋,得質(zhì)量濃度為0.500 0 μg/mL的內(nèi)標(biāo)工作溶液。
2.3 大黃酸校正標(biāo)樣和質(zhì)控血漿樣品的配制及處理
精密吸取空白血漿200 μL,轉(zhuǎn)移至1.5 mL離心管中,加入大黃酸標(biāo)準(zhǔn)曲線、質(zhì)控工作溶液或定量下限工作溶液10 μL和內(nèi)標(biāo)工作溶液50 μL,再加入蛋白沉淀劑(甲醇)600 μL,振蕩混勻約1 min,于4℃下以23 755×g離心10 min。向潔凈進(jìn)樣瓶中加入稀釋劑(水)600 μL,再加入上述離心后的上清液300 μL,混勻,備測(cè)。
2.4 受試者待測(cè)血漿樣品的處理
精密吸取受試者待測(cè)血漿樣品200 μL,轉(zhuǎn)移至1.5 mL離心管中,加入甲醇10 μL(用于體積補(bǔ)償)和內(nèi)標(biāo)工作溶液50 μL,其余步驟按“2.3”項(xiàng)下方法操作,備測(cè)。
2.5 選擇性試驗(yàn)
分別取空白血漿200 μL按“2.3”項(xiàng)下方法處理(不加內(nèi)標(biāo))后所得樣品、空白血漿200 μL與大黃酸標(biāo)準(zhǔn)曲線工作溶液(20.00 μg/mL)10 μL混勻后按“2.3”項(xiàng)下方法處理所得樣品、某受試者餐后口服雙醋瑞因膠囊(參比制劑)50 mg后1.0 h的血漿樣品按“2.4”項(xiàng)下方法處理所得樣品適量,按“2.1”項(xiàng)下條件進(jìn)樣分析,記錄色譜圖(圖1,空白血漿樣品圖略)。結(jié)果顯示,大黃酸、內(nèi)標(biāo)的保留時(shí)間分別約為2.4、3.6 min,內(nèi)源性物質(zhì)不干擾大黃酸和內(nèi)標(biāo)的測(cè)定。
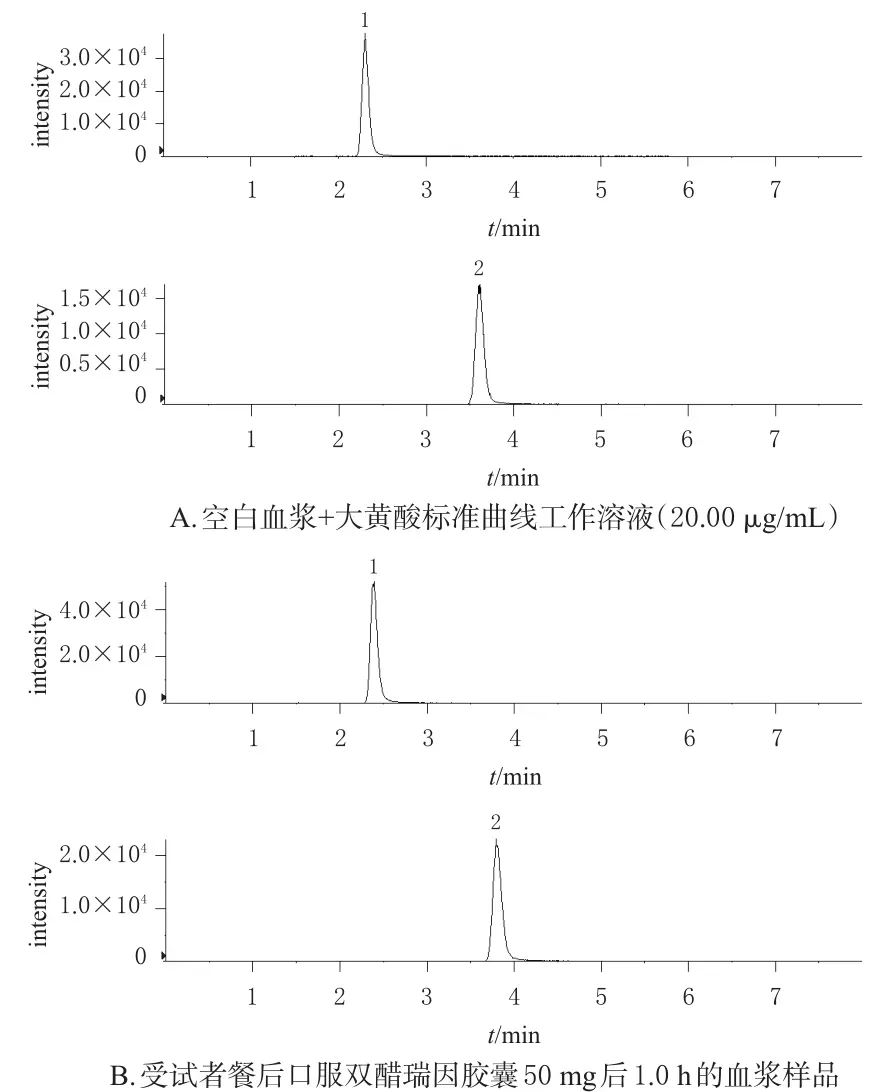
圖1 大黃酸定量分析的典型MRM圖
2.6 標(biāo)準(zhǔn)曲線繪制和定量下限考察
按“2.3”項(xiàng)下方法制得大黃酸質(zhì)量濃度分別為30.00、100.0、200.0、500.0、1 000、2 000、5 000、10 000 ng/mL的校正標(biāo)樣并進(jìn)行處理后,再按“2.1”項(xiàng)下條件進(jìn)樣分析,記錄峰面積。以待測(cè)物質(zhì)量濃度為橫坐標(biāo)(c)、待測(cè)物與內(nèi)標(biāo)的峰面積比值為縱坐標(biāo)(y),采用最小二乘法(權(quán)重系數(shù)為1/c2)進(jìn)行線性回歸,得回歸方程為y=0.001 4c+0.012 5(r=0.999 4),大黃酸檢測(cè)質(zhì)量濃度的線性范圍為30.00~10 000ng/mL,定量下限為30.00ng/mL。
2.7 殘留試驗(yàn)
取3個(gè)空白血漿樣品和“2.6”項(xiàng)下最高質(zhì)量濃度校正標(biāo)樣(10 000 ng/mL)進(jìn)行殘留試驗(yàn)。在檢測(cè)最高質(zhì)量濃度校正標(biāo)樣后連續(xù)檢測(cè)3個(gè)空白血漿樣品,記錄峰面積。結(jié)果顯示,3個(gè)空白血漿樣品中大黃酸和內(nèi)標(biāo)的峰面積均為0,提示殘留效應(yīng)在可接受范圍內(nèi),不影響待測(cè)物的檢測(cè)[5]。
2.8 精密度與準(zhǔn)確度試驗(yàn)
按“2.3”項(xiàng)下方法制得大黃酸質(zhì)量濃度為30.00 ng/mL的定量下限質(zhì)控血漿樣品和80.00、800.0、8 000 ng/mL的低、中、高質(zhì)量濃度質(zhì)控血漿樣品。在至少2 d內(nèi),使用3個(gè)獨(dú)立分析批、4個(gè)質(zhì)量濃度的質(zhì)控血漿樣品(每質(zhì)量濃度平行6份)來評(píng)估批內(nèi)、批間精密度(以RSD表示)和準(zhǔn)確度(以RE表示),結(jié)果見表1。

表1 大黃酸定量分析的精密度與準(zhǔn)確度試驗(yàn)結(jié)果(n=6)
2.9 提取回收率試驗(yàn)
按“2.3”項(xiàng)下方法制得大黃酸質(zhì)量濃度分別為80.00、800.0、8 000 ng/mL的低、中、高質(zhì)量濃度質(zhì)控血漿樣品(每質(zhì)量濃度平行6份)并進(jìn)行處理后,再按“2.1”項(xiàng)下條件進(jìn)樣分析,得大黃酸和內(nèi)標(biāo)的峰面積(A1、A2)。精密吸取空白血漿200 μL,按“2.3”項(xiàng)下方法處理后,取上清液,加入大黃酸質(zhì)控工作溶液,使其質(zhì)量濃度與低、中、高質(zhì)量濃度對(duì)應(yīng)(每質(zhì)量濃度平行6份),再按“2.1”項(xiàng)下條件進(jìn)樣分析,得大黃酸和內(nèi)標(biāo)的峰面積(A3、A4)。分別以A1/A3×100%、A2/A4×100%計(jì)算大黃酸和內(nèi)標(biāo)的提取回收率,結(jié)果見表2。

表2 大黃酸定量分析的提取回收率與基質(zhì)效應(yīng)試驗(yàn)結(jié)果(n=6)
2.10 基質(zhì)效應(yīng)試驗(yàn)
精密吸取6個(gè)不同來源的空白血漿200 μL,分別置于1.5 mL離心管中,加入蛋白沉淀試劑(甲醇)600 μL,振蕩混勻約1 min,于2~8℃下以23 755×g離心10 min,取上清液,即得血漿基質(zhì)樣品。精密吸取水200 μL,置于1.5 mL離心管中,按上述方法處理,得純?nèi)芤悍腔|(zhì)樣品。取血漿基質(zhì)樣品或純?nèi)芤悍腔|(zhì)樣品200 μL,加入大黃酸質(zhì)控工作溶液10 μL和內(nèi)標(biāo)工作溶液50 μL,混勻,即得低、中、高質(zhì)量濃度樣品溶液(每質(zhì)量濃度平行6份)。取上述溶液300 μL,加入稀釋劑(水)600 μL,混勻后,分別按“2.1”項(xiàng)下條件進(jìn)樣分析,記錄大黃酸峰面積(B血漿基質(zhì)、B非基質(zhì))和內(nèi)標(biāo)峰面積(C血漿基質(zhì)、C非基質(zhì)),分別以B血漿基質(zhì)/B非基質(zhì)×100%、C血漿基質(zhì)/C非基質(zhì)×100%計(jì)算大黃酸和內(nèi)標(biāo)的基質(zhì)因子,結(jié)果見表2。以大黃酸基質(zhì)因子除以內(nèi)標(biāo)基質(zhì)因子,計(jì)算得低、中、高質(zhì)量濃度樣品溶液中大黃酸的平均內(nèi)標(biāo)歸一化基質(zhì)因子分別為96.2%、94.7%、92.4%。
2.11 穩(wěn)定性試驗(yàn)
將80.00、800.0、8 000 ng/mL的低、中、高質(zhì)量濃度質(zhì)控血漿樣品(每質(zhì)量濃度平行3份)在室溫白光下放置24 h、-20℃下儲(chǔ)存49 d和反復(fù)凍融(-20℃~室溫)循環(huán)3次后,再按“2.3”項(xiàng)下方法處理,測(cè)得各樣品實(shí)測(cè)質(zhì)量濃度與理論質(zhì)量濃度的RE均在±10.0%以內(nèi),提示質(zhì)控血漿樣品在上述條件下放置的穩(wěn)定性較好。將低、中、高質(zhì)量濃度質(zhì)控血漿樣品按“2.3”項(xiàng)下方法處理后,在自動(dòng)進(jìn)樣器(4℃)中放置23 h或在室溫白光下放置24 h,測(cè)得各樣品實(shí)測(cè)質(zhì)量濃度與理論質(zhì)量濃度的RE均在±10.0%以內(nèi),提示經(jīng)處理的質(zhì)控血漿樣品在上述條件下放置的穩(wěn)定性較好。將大黃酸和內(nèi)標(biāo)貯備溶液(質(zhì)量濃度均為1.0 mg/mL)在室溫白光下放置24 h或冷藏(2~8℃)保存47 d,測(cè)得各溶液實(shí)測(cè)質(zhì)量濃度與理論質(zhì)量濃度的RE均在±10.0%以內(nèi),提示各貯備溶液在上述條件下放置的穩(wěn)定性較好。
2.12 兩種制劑生物等效性評(píng)價(jià)
2.12.1 研究對(duì)象 本研究方案經(jīng)蘇州大學(xué)附屬第二醫(yī)院醫(yī)學(xué)倫理委員會(huì)審核批準(zhǔn)[倫理批件號(hào)為(2014)倫審第(22)號(hào)]。受試者入選標(biāo)準(zhǔn)包括:>18周歲,體質(zhì)量指數(shù)(BMI)為19.0~26.0 kg/m2,試驗(yàn)前14 d病史、生命體征、體格檢查,實(shí)驗(yàn)室檢查及篩選期其他相關(guān)檢查均正常或未見有臨床意義的異常。排除標(biāo)準(zhǔn)包括:過敏體質(zhì)或有藥物、食物過敏史者;篩選前12個(gè)月內(nèi)有嗜煙(每天吸煙數(shù)量≥5支)史者;篩選前12個(gè)月內(nèi)有藥物濫用史或成癮性物質(zhì)檢測(cè)陽性者;篩選前3個(gè)月內(nèi)接受過手術(shù)(特別是會(huì)影響藥物吸收、分布、代謝、排泄的手術(shù))或者計(jì)劃在試驗(yàn)期間進(jìn)行手術(shù)者;篩選前14 d內(nèi)有處方藥、非處方藥、中草藥、保健品服用史者。按上述標(biāo)準(zhǔn),本研究最終納入健康受試者24名。所有受試者均簽署知情同意書,年齡為(24±3)歲,身高為(1.70±0.06)m,體質(zhì)量為(61.9±6.9)kg,BMI符合上述標(biāo)準(zhǔn)。
2.12.2 試驗(yàn)設(shè)計(jì) 本研究采用隨機(jī)、開放、雙周期交叉試驗(yàn)設(shè)計(jì):將24名受試者隨機(jī)分成2組(每組12名),禁食過夜10 h后,于每周期試驗(yàn)首日早晨進(jìn)食標(biāo)準(zhǔn)餐30 min后,分別口服受試制劑或參比制劑50 mg(用溫開水240 mL送服)。受試者兩周期服藥順序由隨機(jī)結(jié)果決定,清洗期為1周。分別于服藥前以及服藥后20、40 min和1.0、1.5、2.0、2.5、3.0、4.0、5.0、6.0、7.0、8.0、10、12、15、24 h時(shí)于肘靜脈取血3 mL,置于肝素鈉采血管中,于4℃下以2 304×g離心5 min,分離上層血漿,將血漿分成2份(1份用于檢測(cè),1份用于備份),于-20℃下凍存。本研究所用標(biāo)準(zhǔn)餐食譜符合餐后生物等效性研究設(shè)計(jì)中對(duì)高脂高熱量食譜的要求[6]。
2.12.3 生物等效性評(píng)價(jià) 24名受試者均完成試驗(yàn)。取其凍存的血漿樣品,按“2.4”項(xiàng)下方法處理后,再按“2.1”項(xiàng)下條件進(jìn)樣分析,記錄峰面積,以隨行標(biāo)準(zhǔn)曲線計(jì)算血漿樣品中大黃酸的質(zhì)量濃度。結(jié)果顯示,受試者血漿樣品中大黃酸的質(zhì)量濃度為35~7 305 ng/mL,均在線性范圍內(nèi)。采用DAS 3.2.9軟件繪制平均血藥濃度-時(shí)間曲線并計(jì)算主要藥動(dòng)學(xué)參數(shù)。藥動(dòng)學(xué)參數(shù)包括血藥濃度-時(shí)間曲線下面積(AUC0-24h、AUC0-∞)、峰濃度(cmax)、達(dá)峰時(shí)間(tmax)、半衰期(t1/2)。其中,cmax和tmax均以實(shí)測(cè)值表示;AUC0-24h用梯形法計(jì)算;AUC0-∞和t1/2分別按下式計(jì)算:AUC0-∞=AUC0-24h+ct/λz,t1/2=0.693/λz(式中,ct為最后1個(gè)時(shí)間點(diǎn)的血藥濃度;λz為末端消除速率常數(shù),以對(duì)數(shù)血藥濃度-時(shí)間曲線末端直線部分的斜率求得)。24名受試者餐后口服受試制劑和參比制劑50 mg后,大黃酸的平均血藥濃度-時(shí)間曲線見圖2,主要藥動(dòng)學(xué)參數(shù)見表3。
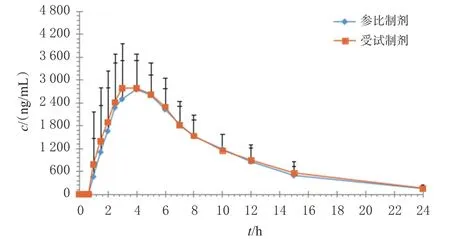
圖2 受試者餐后口服受試制劑和參比制劑50 mg后大黃酸的平均血藥濃度-時(shí)間曲線(n=24)
表3 受試者餐后口服受試制劑和參比制劑50 mg后大黃酸的主要藥動(dòng)學(xué)參數(shù)(±s,n=24)

表3 受試者餐后口服受試制劑和參比制劑50 mg后大黃酸的主要藥動(dòng)學(xué)參數(shù)(±s,n=24)
a:tmax以“中位數(shù)(最小值,最大值)”表示
制劑受試制劑參比制劑t1/2/h 4.26±1.12 4.19±1.05 AUC0-24 h/(ng·h/mL)25764±6134 24316±5856 AUC0-∞/(ng·h/mL)26679±6409 25170±6415 cmax/(ng/mL)3517±1121 3225±755 tmaxa/h 3.50(0.67,12.00)4.00(1.50,7.00)
本研究對(duì)受試者餐后狀態(tài)下的主要藥動(dòng)學(xué)參數(shù)(cmax、AUC0-24h、AUC0-∞)進(jìn)行自然對(duì)數(shù)轉(zhuǎn)換,然后以多因素方差分析進(jìn)行顯著性檢驗(yàn),用雙向單側(cè)t檢驗(yàn)及90%置信區(qū)間來評(píng)價(jià)受試制劑與參比制劑的生物等效性。若經(jīng)對(duì)數(shù)轉(zhuǎn)換后受試制劑和參比制劑cmax、AUC0-24h、AUC0-∞幾何均值比的90%置信區(qū)間落在接受范圍(80.00%~125.00%)之內(nèi),則表示兩制劑生物等效[5]。對(duì)tmax進(jìn)行配對(duì)秩和檢驗(yàn)。采用DAS 3.2.9軟件進(jìn)行上述統(tǒng)計(jì)分析,檢驗(yàn)水準(zhǔn)α=0.05。
方差分析模型中,以序列、藥物、周期為固定效應(yīng),受試者(序列)為隨機(jī)效應(yīng),計(jì)算主要藥動(dòng)學(xué)參數(shù)幾何均值比(受試制劑/參比制劑)的90%置信區(qū)間。結(jié)果顯示,兩制劑cmax、AUC0-24h、AUC0-∞幾何均值比的90%置信區(qū)間分別為 100.8%~113.9%、103.1%~109.4%、103.2%~109.9%,相對(duì)生物利用度(受試制劑與參比制劑AUC0-24h之比)為(106.6±8.9)%。由此可判定,在餐后狀態(tài)下,兩制劑生物等效。此外,受試制劑的tmax與參比制劑比較,差異無統(tǒng)計(jì)學(xué)意義(P>0.05)。
在試驗(yàn)過程中,所有受試者均未見明顯不良事件發(fā)生。相關(guān)檢測(cè)結(jié)果顯示,24名受試者血常規(guī)、尿常規(guī)等檢測(cè)值異常發(fā)生率低,且異常并無臨床意義;同時(shí),未見有臨床意義的心電圖變化,其收縮壓、舒張壓、脈搏、體溫均在正常范圍內(nèi)波動(dòng)。
3 討論
血漿中大黃酸濃度的測(cè)定方法包括高效液相色譜-紫外光譜(HPLC-UV)法[7-8]和 HPLC-MS/MS 法[4,9-10]。近年來,血漿大黃酸濃度的測(cè)定以靈敏度更高、專屬性更強(qiáng)的HPLC-MS/MS法為主。本研究樣本量較大,甲醇直接沉淀蛋白法具有簡便、快捷的優(yōu)點(diǎn)[11],可滿足雙醋瑞因膠囊藥動(dòng)學(xué)研究的大樣本檢測(cè)。因此,本研究采用甲醇沉淀蛋白進(jìn)行血漿樣品前處理,定量下限為30.00 ng/mL,低于文獻(xiàn)所示結(jié)果[4]。
在血藥濃度測(cè)定方面,本課題組對(duì)液相條件進(jìn)行了優(yōu)化:在水相中加入醋酸銨,有利于大黃酸和內(nèi)標(biāo)色譜峰峰形的改善。雖然有研究指出,在負(fù)離子掃描模式下,若在水相中加入酸性物質(zhì)(如甲酸),理論上會(huì)減弱大黃酸的色譜響應(yīng)[12]。但本課題組前期考察結(jié)果顯示,在水相中加入少量甲酸有利于大黃酸和內(nèi)標(biāo)色譜峰峰形的進(jìn)一步改善,且不會(huì)影響大黃酸的定量下限;同時(shí),當(dāng)在水相中加入0.4%的甲酸時(shí),大黃酸色譜峰的響應(yīng)較強(qiáng)且保留時(shí)間適中。此外,筆者在檢測(cè)方法建立的過程中發(fā)現(xiàn),在等度洗脫條件下連續(xù)分析多個(gè)樣品后,大黃酸的質(zhì)譜響應(yīng)波動(dòng)明顯且有減弱趨勢(shì),分析原因可能與蛋白沉淀所得血漿樣品中內(nèi)源性雜質(zhì)較多有關(guān),遂在保證試驗(yàn)效率的基礎(chǔ)上,采取如下措施:(1)在沉淀蛋白后,取上清液300 μL并加水600 μL稀釋,混勻后再進(jìn)樣分析,有助于減少基質(zhì)效應(yīng)的干擾并降低液質(zhì)聯(lián)用儀、色譜柱被污染的可能性;(2)將流動(dòng)相洗脫方式優(yōu)化為梯度洗脫,即先用75%甲醇等度洗脫,使大黃酸及內(nèi)標(biāo)出峰;再用95%甲醇洗脫,洗去內(nèi)源性雜質(zhì)以減少對(duì)后續(xù)樣品檢測(cè)的影響。
本研究報(bào)道了餐后狀態(tài)下中國健康受試者口服雙醋瑞因膠囊后主要代謝產(chǎn)物大黃酸的藥動(dòng)學(xué)數(shù)據(jù)。Mandawgade等[13]報(bào)道,印度健康志愿者餐后口服雙醋瑞因膠囊50 mg,其體內(nèi)大黃酸的AUC0-∞、cmax、tmax分別為26 578.63 ng·h/mL、4 298.63 ng/mL、5 h;Nicolas等[14]報(bào)道,法國健康志愿者餐后口服雙醋瑞因膠囊50 mg后,其體內(nèi)大黃酸的AUC0-∞為25 900 ng·h/mL,tmax為5.2 h。本研究所得受試制劑和參比制劑的主要藥動(dòng)學(xué)參數(shù)AUC0-∞、tmax與上述文獻(xiàn)接近,但cmax與Mandawgade等[13]的結(jié)果有所差異,這可能與樣本量和受試者人群不同有關(guān)。本研究結(jié)果顯示,受試制劑(國產(chǎn)雙醋瑞因膠囊)和參比制劑(安必丁?)生物等效。
綜上所述,在受試者餐后狀態(tài)下,受試制劑和參比制劑生物等效。本研究采用甲醇沉淀蛋白預(yù)處理血漿樣品,使用HPLC-MS/MS法測(cè)定了中國健康受試者餐后口服雙醋瑞因膠囊后血漿中活性代謝產(chǎn)物大黃酸的濃度,計(jì)算了該成分的主要藥動(dòng)學(xué)數(shù)據(jù),并評(píng)價(jià)了受試制劑與參比制劑的生物等效性,為國產(chǎn)雙醋瑞因膠囊上市及臨床應(yīng)用提供了參考依據(jù)。
猜你喜歡
現(xiàn)代臨床醫(yī)學(xué)(2022年4期)2022-09-29 07:38:00
中學(xué)生數(shù)理化·八年級(jí)物理人教版(2021年12期)2021-12-31 03:23:08
昆明醫(yī)科大學(xué)學(xué)報(bào)(2021年4期)2021-07-23 01:21:50
中學(xué)生數(shù)理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
云南醫(yī)藥(2019年3期)2019-07-25 07:25:14
產(chǎn)品可靠性報(bào)告(2017年7期)2017-09-05 09:49:12
海南醫(yī)學(xué)(2016年8期)2016-06-08 05:43:00
汽車觀察(2016年3期)2016-02-28 13:16:26
醫(yī)學(xué)研究雜志(2015年9期)2015-07-01 17:28:15
