我國附條件批準上市抗腫瘤藥物探討*
2022-10-11 01:21:42宋茵茵徐文峰金鵬飛
中國藥業 2022年19期
宋茵茵,徐文峰,胡 欣,金鵬飛△
(1.北京醫院藥學部·國家老年醫學中心·中國醫學科學院老年醫學研究院·北京市藥物臨床風險與個體化應用評價重點實驗室<北京醫院>,北京 100730;2.北京大學藥學院藥事管理與臨床藥學系,北京 100191)
藥品附條件批準上市是指針對具有突出臨床價值的急需藥品,已有臨床研究數據,雖尚未滿足常規上市注冊的全部要求,但藥物臨床試驗已有數據證實療效,并能預測其臨床價值,為滿足臨床和患者的醫療急需,在規定申請人必須履行特定條件的情況下,基于替代終點、中間臨床終點或早期臨床試驗數據而批準上市。目前,中國支持治療嚴重危及生命且尚無有效治療手段疾病的藥品(如抗腫瘤藥物等)、公共衛生方面急需藥品及應對重大突發公共衛生事件急需疫苗等藥品的附條件批準[1]。附條件批準、突破性治療藥物、優先審評審批及特別審批程序共同組成了我國藥品加快上市注冊程序的4種批準通道[2],上述通道既滿足了醫療急需,又為研發機構提供了在市場競爭中獲得領先優勢的契機,可促進以臨床價值為導向的藥品研發、新藥上市和行業創新。《全國第三次死因回顧抽樣調查報告》顯示,我國惡性腫瘤是第二死亡原因,僅次于腦血管疾病,且兩者死亡率十分接近。2015年,我國約有392.9萬例新發惡性腫瘤患者,年齡標準化發病率和死亡率分別為每10萬人口186.39和105.84[3]。由于腫瘤疾病的特殊性,評估抗腫瘤藥物有效性和安全性的臨床研究也具有一定的復雜性和長期性。抗腫瘤藥物上市的常規批準程序耗時長、成本高,但患者的生存期較短,對抗腫瘤藥物的治療等待期有限。附條件批準程序有助于抗腫瘤藥物的快速上市,使通過獲益風險評估的抗腫瘤藥物提前用于臨床急需患者,從而提高抗腫瘤藥物的可及性。我國自附條件批準程序落地實施以來,已有多種抗腫瘤藥物獲批上市。本研究中收集了國家藥品監督管理局藥品審評中心(CDE)已公開的附條件批準上市抗腫瘤藥物上市技術審評報告,梳理了藥品監管機構公開的藥品審評信息,總結了國內抗腫瘤藥物附條件批準上市品種和所附條件,為抗腫瘤藥物的審批和合理用藥提供參考。現報道如下。
1 我國附條件批準相關制度依據和技術要求
1.1 有條件批準
2016年2月發布的《關于解決藥品注冊申請積壓實行優先審評審批的意見》(已廢止)提出,對治療嚴重危及生命且尚無有效治療手段的疾病,以及解決臨床需求具有重大意義的新藥,若根據早期臨床試驗數據,可合理預測或判斷其臨床獲益且較現有治療手段具有明顯優勢,允許在完成Ⅲ期確證性臨床試驗前有條件批準上市[4]。首次在我國引入藥品不完全批準上市這一概念。
1.2 附條件批準
2017年10月發布的《關于深化審評審批制度改革鼓勵藥品醫療器械創新的意見》提出,加快臨床急需藥品醫療器械的審評審批。對治療嚴重危及生命且尚無有效治療手段的疾病及公共衛生方面等急需的藥品、醫療器械,臨床試驗早期、中期指標顯示療效并可預測其臨床價值的,可附帶條件批準上市[5]。這是我國首次使用“附條件批準”來表示藥品醫療器械的不完全批準上市。
2019年修訂并實施的《中華人民共和國藥品管理法》《中華人民共和國疫苗管理法》首次在法律法規層面明確了“附條件批準”,即:“對治療嚴重危及生命且尚無有效治療手段的疾病以及公共衛生方面急需的藥品,藥物臨床試驗已有數據顯示療效并能預測其臨床價值的,可以附條件批準,并在藥品注冊證書中載明相關事項。”[2]
2020年1月修訂并于7月實施的《藥品注冊管理辦法》中,對附條件批準程序的適用范圍、申請程序、上市后要求及終止程序進行了詳細規定[6]。后續發布的《藥品附條件批準上市申請審評審批工作程序(試行)》《藥品附條件批準上市技術指導原則(試行)》進一步明確了藥品附條件批準上市申請審評審批的工作程序[7]和附條件批準上市的具體技術要求[1]。上述法規制度、工作程序和技術要求共同構成了我國的藥品附條件批準上市程序,為加快我國符合特定條件的臨床急需藥品批準上市提供了強有力的制度和技術支持。詳見表1。
2 國內抗腫瘤藥物附條件批準上市現狀
截至2022年2月,CDE已公開15個抗腫瘤藥物、涉及19個適應證的附條件批準上市審評信息(表2至表16),所有藥品的技術審評報告均注明需要在上市后繼續完善臨床研究方面的內容,主要集中在開展上市后臨床研究和嚴格執行上市后風險管理計劃等方面。其中,藥品上市后風險管理計劃的安全性監測和風險控制指標包括前期臨床或非臨床數據提示的重要的已識別風險、重要的潛在風險,以及特殊人群用藥的安全性、藥物相互作用信息等尚無相關數據提示的重要的缺失信息。不同抗腫瘤藥物的指標選擇不同,取決于藥物本身復雜的風險機制。如奧布替尼重要的已識別風險主要指出血風險,重要的潛在風險包括血細胞減少癥、感染、心律失常、高血壓等,重要的缺失信息包括不同特殊人群安全性、藥物相互作用信息等;替雷利珠單抗重要的已識別風險包括溶血性輸血反應、輸液相關反應和免疫相關事件,重要的潛在風險包括迄今未報告的其他自身免疫類疾病和對發育中嬰兒的影響(胚胎-胎兒毒性),重要的缺失信息主要指不同特殊人群的安全性信息。此外,少數藥品的藥學研究信息也需上市后補充完善。如西達帕胺需結合生產經驗,制訂固體分散體的粒度及粒度分布標準,以保證制劑的含量均勻度;此外,相對于5 mg的規格,確定的臨床用量為每次30 mg,建議后續結合臨床治療經驗的積累,設計和開發更方便的臨床用藥規格。阿美替尼的商業生產如需放大批量,應注意開展相應的驗證研究,必要時應針對生產規模放大提出補充申請等。

表1 我國藥品附條件批準上市相關制度文件與內容Tab.1 Documents and content of relevant systems of conditional approval for marketing of drugs in China

表2 西達本胺片:CDE附條件批準上市審評審批信息Tab.2 Chidamide Tablets:review and approval information of the conditional approval for marketing released by the CDE

表16 達雷妥尤單抗注射液:CDE附條件批準上市審評審批信息Tab.16 Daratumumab Injection:review and approval information of the conditional approval for marketing released by the CDE
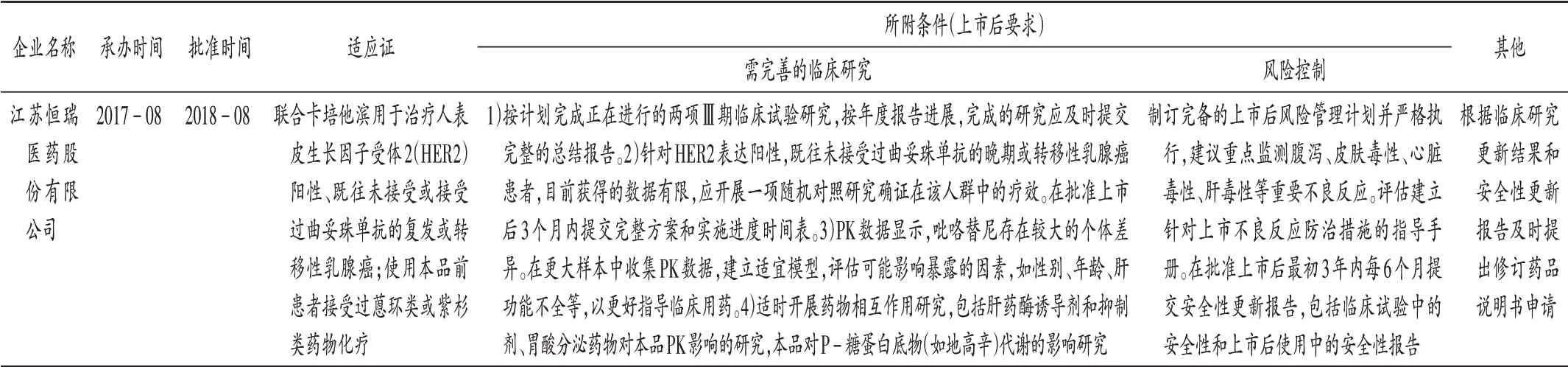
表3 馬來酸吡咯替尼片:CDE附條件批準上市審評審批信息Tab.3 Pyrotinib Maleate Tablets:review and approval information of the conditional approval for marketing released by the CDE

表4 安羅替尼膠囊:CDE附條件批準上市審評審批信息Tab.4 Anlotinib Capsules:review and approval information of the conditional approval for marketing released by the CDE

表5 澤布替尼膠囊:CDE附條件批準上市審評審批信息Tab.5 Zanubrutinib Capsules:review and approval information of the conditional approval for marketing released by the CDE

表6 奧布替尼片:CDE附條件批準上市審評審批信息Tab.6 Orelabrutinib Tablets:review and approval information of the conditional approval for marketing released by the CDE
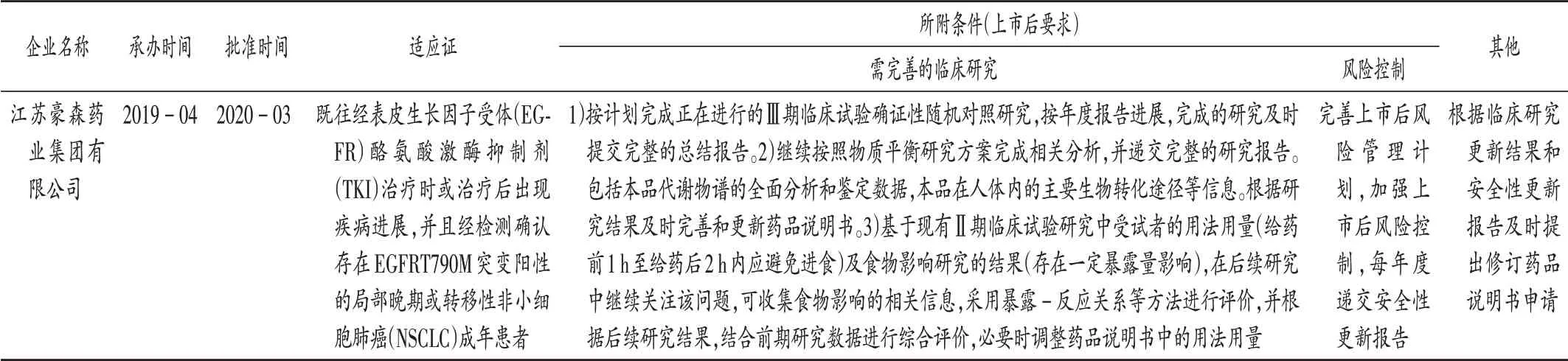
表7 甲磺酸阿美替尼片:CDE附條件批準上市審評審批信息Tab.7 Almonertinib Mesilate Tablets:review and approval information of the conditional approval for marketing released by the CDE

表8 氟唑帕利膠囊:CDE附條件批準上市審評審批信息Tab.8 Fluzoparib Capsules:review and approval information of the conditional approval for marketing released by the CDE

表9 甲苯磺酸尼拉帕利膠囊:CDE附條件批準上市審評審批信息Tab.9 Niraparib Tosilate Capsules:review and approval information of the conditional approval for marketing released by the CDE

表10 替雷利珠單抗注射液:CDE附條件批準上市審評審批信息Tab.10 Tislelizumab Injection:review and approval information of the conditional approval for marketing released by the CDE

表11 特瑞普利單抗注射液:CDE附條件批準上市審評審批信息Tab.11 Toripalimab Injection:review and approval information of the conditional approval for marketing released by the CDE
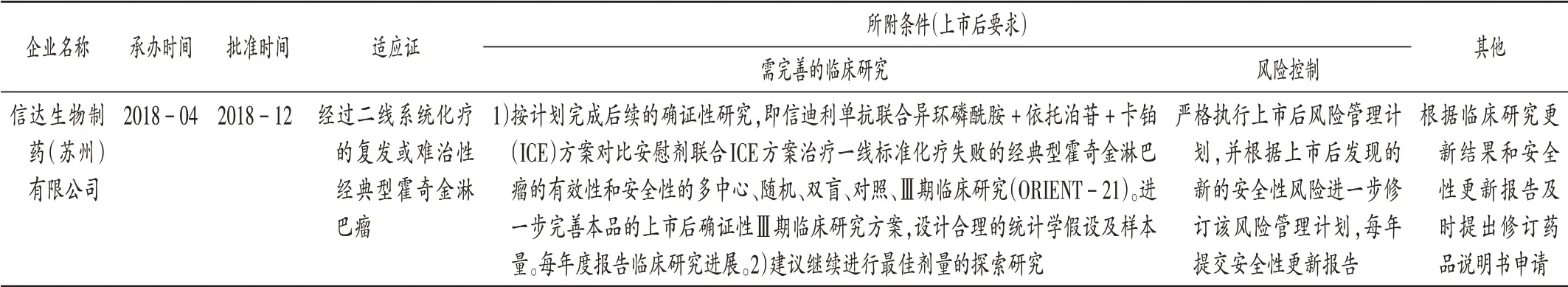
表12 信迪利單抗注射液:CDE附條件批準上市審評審批信息Tab.12 Sintilimab Injection:review and approval information of the conditional approval for marketing released by the CDE

表13 注射用卡瑞利珠單抗:CDE附條件批準上市審評審批信息Tab.13 Camrelizumab for Injection:review and approval information of the conditional approval for marketing released by the CDE

表14 普拉曲沙注射液:CDE附條件批準上市審評審批信息Tab.14 Pralatrexate Injection:review and approval information of the conditional approval for marketing released by the CDE

表15 帕米帕利膠囊:CDE附條件批準上市審評審批信息Tab.15 Pamiparib Capsules:review and approval information of the conditional approval for marketing released by the CDE
3 討論
附條件批準上市藥品的藥學、藥理毒理學要求與常規批準上市藥品相同,與常規批準上市藥品的主要區別為可基于替代終點、中間臨床終點或早期臨床試驗數據批準上市。根據《藥品附條件批準上市技術指導原則(試行)》中的技術要求,附條件批準上市后要求應至少包括上市后臨床研究計劃、研究完成日期、最終臨床研究報告提交日期、上市后風險管理計劃等內容,申請人應承諾按時完成所有的臨床試驗[1]。
抗腫瘤藥物的附條件批準上市一般是基于以治療作用探索而非治療作用確證為目的的Ⅱ期臨床試驗結果。相比于完全批準上市的藥品,其終點數據尚未獲得,臨床數據鏈尚不完善,有效性和安全性仍需意義更明確的證據支持。有效性評價方面,腫瘤作為一種特殊的慢性疾病,其治療目標為控制疾病進展、改善生活質量或延長生存時間。其中,延長總生存期是腫瘤治療的終極目標[8]。附條件批準基于的早期臨床研究往往人群暴露量小,且采用具有不確定性的替代終點指標,因此,即使研究取得了陽性結果,其證據等級一般較低,有效性結果穩健性有限,仍不能完全確證抗腫瘤藥物在患者中應用的真實療效。安全性評價方面,由于抗腫瘤藥物的特殊性,其自身不良反應風險一般高于非抗腫瘤藥物。受限于早期臨床研究周期短、人群暴露量小等,部分不良反應尤其是長期安全性相關的不良反應未能及時發現和報告。因此,對于能為臨床帶來顯著獲益的抗腫瘤藥物,盡管附條件批準上市程序極大地縮短了其從藥品研發到批準上市的時間間隔,提高了藥品的可及性,但同時也給臨床用藥帶來了一定挑戰。需要申請人開展設計更嚴謹、結局更具臨床價值的上市后臨床研究(如Ⅲ期確證性隨機對照試驗),證實抗腫瘤藥物給患者帶來的長期臨床獲益和長期安全性結果,以完善臨床數據鏈。此外,從保證患者用藥安全的角度考慮,也需要申請人制訂完備的上市后風險管理計劃,明確已存在或已識別的風險及潛在風險,并按該計劃加強藥品上市后安全性監測和風險控制。
本研究中通過整理CDE公開的抗腫瘤藥物的上市審評信息發現,對于上述附條件批準上市的抗腫瘤藥物,其上市所附條件均涉及上市后臨床研究計劃和上市后風險管理計劃,其中上市后臨床研究的具體要求結合其特點略有不同。申請人需提交確定的研究計劃和方案,并開展新的臨床試驗,或繼續按計劃完成正在進行的臨床試驗。這些臨床試驗通常是以確認預期的臨床獲益為目的的上市后確證性臨床試驗,可為藥品上市的完全批準提供充足證據。此外,對于某些抗腫瘤藥物,附條件批準也要求申請人上市后開展在特殊人群中的藥代動力學和用法用量研究、物質平衡研究、最佳劑量探索研究、藥物-藥物相互作用研究、食物對生物利用度的影響研究等。對于上市后風險管理,申請人需提交相對完善的上市后風險管理計劃,并在上市后嚴格執行該計劃,定期遞交關于臨床試驗中的安全性和上市后使用中的安全性的藥品安全性更新報告。但多數抗腫瘤藥物附條件批準所附條件中未明確上市后研究完成日期和最終臨床研究報告提交日期,僅要求按年度報告臨床研究進展,并在研究完成后及時提交完整的總結報告。僅氟唑帕利膠囊、替雷利珠單抗注射液、注射用卡瑞利珠單抗、帕米帕利膠囊等少數藥物明確上市后研究完成的期限,即批準上市后3年內或5年內完成研究,并以補充申請形式遞交完整的研究總結報告,以支持完全批準。
近年來,隨著一系列法規制度、工作程序和技術要求的發布與實施,我國藥品附條件批準程序逐漸完善。經多年實踐,我國附條件批準程序取得了一定成效,積累了一定經驗,目前已有多個抗腫瘤藥物通過附條件批準上市。我國附條件批準上市制度與國際基本接軌[9-10],但參考國外附條件批準上市程序,仍可完善相應的實施細則,提出更明確、更具體的技術要求,如根據附條件批準上市藥物的不同屬性,對上市后臨床研究計劃及上市后風險管理計劃的具體內容給出明確建議,為我國抗腫瘤藥物附條件批準程序更好地開展提供參考。此外,在技術審評方面,仍需要一些通用、規范的技術指南或共識支持附條件批準上市的技術審評,并引導藥品研發企業正確認識附條件批準的制度要求和具體內涵,重視附條件批準上市的上市后要求,從而保障我國抗腫瘤藥物附條件批準上市程序的規范實施,促進其健康、長遠和科學發展,進而有利于更好地解決臨床用藥的急需問題,保障腫瘤患者用藥的可及性。
猜你喜歡
車主之友(2022年6期)2023-01-30 08:01:04
車主之友(2022年4期)2022-11-25 07:27:30
車主之友(2022年4期)2022-08-27 00:57:48
現代儀器與醫療(2022年2期)2022-08-11 09:51:40
汽車工程師(2021年12期)2022-01-18 06:02:43
人大建設(2019年12期)2019-05-21 02:55:44
瞭望東方周刊(2017年42期)2017-12-05 18:49:38
環球時報(2017-03-30)2017-03-30 06:44:45
信息安全與通信保密(2016年3期)2016-08-23 01:23:46
中國衛生(2015年3期)2015-11-19 02:53:32
