高效液相色譜法測(cè)定蜂蜜中諾氟沙星殘留
2022-10-13 07:41:54魏超田高倩妮
食品安全導(dǎo)刊 2022年25期
魏超田,高倩妮
(1.安徽正鑒檢驗(yàn)檢測(cè)有限公司,安徽亳州 236800;2.安徽萬花草生物科技有限公司,安徽亳州 236800;3.亳州學(xué)院,安徽亳州 236800)
隨著人們對(duì)健康和天然保健產(chǎn)品的追求和崇尚,具有抗氧化性、抗菌性等生物活性功能的蜂蜜及其產(chǎn)品的質(zhì)量受到了人們的廣泛關(guān)注。蜂蜜在養(yǎng)殖過程中會(huì)感染疾病或受到蟲害,峰農(nóng)為控制病蟲害的發(fā)生而濫用抗生素,導(dǎo)致農(nóng)藥殘留問題的發(fā)生。諾氟沙星屬喹諾酮類藥物,是在喹諾酮母核的基礎(chǔ)上進(jìn)行人工合成的一類廣譜抗菌藥,因其價(jià)格低、抗菌性強(qiáng),被廣泛用于人和動(dòng)物疾病的治療[1]。但其自身無法分解,在人體內(nèi)會(huì)大量積累殘留,人們食用動(dòng)物組織后可能出現(xiàn)對(duì)該藥物的嚴(yán)重耐藥性。諾氟沙星在蜂蜜養(yǎng)殖過程中屬于禁用藥物,一經(jīng)檢出即為不合格產(chǎn)品[2]。2019年國家市場(chǎng)監(jiān)督管理總局對(duì)蜂蜜抽檢項(xiàng)目中,新增了幾項(xiàng)喹諾酮類,其中諾氟沙星殘留就是其中一項(xiàng)[3]。
目前測(cè)定諾氟沙星殘留量的方法有微生物法、高效液相色譜-紫外或二極管陣列檢測(cè)[4-6]、薄層色譜法[7-9]以及高效液相色譜-串聯(lián)質(zhì)譜法(High Performance Liquid Chromatography Tandem Mass Spectrometry,HPLC-MS/MS))[10-13]。其中質(zhì)譜法運(yùn)用較多,質(zhì)譜法雖然檢出限低但操作復(fù)雜、耗費(fèi)時(shí)間長(zhǎng),且由于設(shè)備條件和成本的限制無法普及。相關(guān)學(xué)者對(duì)諾氟沙星藥物殘留的檢測(cè)方法研究大多集中在畜產(chǎn)品、奶制品、水產(chǎn)品上[14-16],對(duì)于蜂蜜中的諾氟沙星殘留檢測(cè)較少,且動(dòng)物性等產(chǎn)品的前處理方法不適用蜂蜜。本試驗(yàn)通過研究蜂蜜不同的前處理方式,建立了一種簡(jiǎn)單、經(jīng)濟(jì)、高效、實(shí)用性強(qiáng)的高效液相色譜法測(cè)定蜂蜜中的諾氟沙星,以期為快速檢測(cè)蜂蜜中諾氟沙星含量奠定理論基礎(chǔ),為蜂產(chǎn)品質(zhì)量控制提供相應(yīng)保證。
1 材料與方法
1.1 材料與試劑
諾氟沙星標(biāo)準(zhǔn)樣品;乙腈(色譜純),甲酸、乙酸、氫氧化鈉、無水硫酸鎂、磷酸氫二鈉以及磷酸氫二鉀均為分析純,國藥基團(tuán);蜂蜜(5種不同品牌)購自超市。
1.2 儀器與設(shè)備
高效液相色譜儀(配有紫外、熒光檢測(cè)器),賽默飛世爾科技(中國)有限公司;AcclaimTMPolarAdvantage Ⅱ C18柱(250 mm×4.6 mm,5 μm)。
1.3 試驗(yàn)方法
1.3.1 標(biāo)準(zhǔn)曲線的繪制
準(zhǔn)確量取 100 μg·mL-1的諾氟沙星標(biāo)液 10 μL,用0.2%甲酸水溶液定容至1 mL,配制成1.00 μg·mL-1標(biāo)準(zhǔn)儲(chǔ)備液。準(zhǔn)確移取儲(chǔ)備液按梯度稀釋配制成濃度為 0.001 μg·mL-1、0.002 μg·mL-1、0.004 μg·mL-1、0.080 μg·mL-1、0.010 μg·mL-1和 0.200 μg·mL-1的 標(biāo)準(zhǔn)系列工作液,儲(chǔ)備于冰箱中備用。
1.3.2 高效液相色譜條件
色 譜 柱:AcclaimTMPolarAdvantage Ⅱ C18柱(250 mm×4.6 mm,5 μm);熒光檢測(cè)波長(zhǎng):激發(fā)波長(zhǎng)280 nm,發(fā)射波長(zhǎng)450 nm;柱溫:30 ℃;流速:1.5 mL·min-1;進(jìn)樣量 10 μL;流動(dòng)相:流動(dòng)相 A(乙腈)∶流動(dòng)相B(0.2%甲酸水溶液)為13∶87(體積比,下同)。
1.3.3 蜂蜜樣品處理
(1)蜂蜜稀釋液的選擇。①先用水溶解樣品,再用0.5%乙酸的乙腈溶液萃取蜂蜜中的諾氟沙星。②用0.02 mol·L-1氫氧化鈉溶液溶解樣品,再用0.5%乙酸的乙腈溶液萃取蜂蜜中的諾氟沙星。③用Mallvaine pH 4.0緩沖液溶解樣品,再用0.5%乙酸的乙腈溶液提取,最后水浴蒸發(fā)濃縮提取諾氟沙星。
稱取約5.00 g(精確到0.01)蜂蜜,加入5.0 mL 0.02 mol·L-1氫氧化鈉溶液,攪拌均勻,加入25 mL 0.5%乙酸乙腈溶液,渦旋5 min,加入無水硫酸鎂5 g,渦旋2 min,4 000 r·min-1離心5 min,取上清液。
固相萃取柱依次用甲醇、磷酸緩沖鹽各2 mL預(yù)淋,取上清液過柱,10 mL甲醇洗脫,收集液水浴蒸至近干,0.2%乙酸水溶液溶解定容至2 mL,過0.45 μm有機(jī)濾膜,高效液相色譜儀分析測(cè)定。
(2)提取液的選擇。選取0.05 mol·L-1磷酸緩沖鹽溶液、0.5%乙酸的乙腈溶液、乙腈+水(8∶2)混合溶液為蜂蜜中諾氟沙星的提取液,考察不同提取液及其添加量對(duì)蜂蜜中諾氟沙星提取量的影響。
2 結(jié)果與分析
2.1 高效液相色譜方法的建立
2.1.1 流動(dòng)相及其比例的確定
目前高效液相色譜法測(cè)定喹酮類含量時(shí),一般選擇磷酸/三乙胺、磷酸/庚烷磺酸鈉、磷酸/四丁基溴化銨體系作為流動(dòng)相分離喹諾酮類,但這幾種體系中都含有鹽類物質(zhì),易結(jié)晶,測(cè)定中容易造成色譜柱和檢測(cè)器堵塞,極大縮短了色譜柱的使用壽命。
試驗(yàn)中發(fā)現(xiàn)熒光檢測(cè)器對(duì)含有乙腈的流動(dòng)相變化較敏感,且在流動(dòng)相中加入適量的甲酸可以提高其靈敏度,因此本試驗(yàn)在乙腈-0.2%甲酸,0.05 mol·L-1磷酸溶液/三乙胺-乙腈兩種系列作為流動(dòng)相的情況下測(cè)定諾氟沙星,結(jié)果發(fā)現(xiàn)0.05 mol·L-1磷酸溶液/三乙胺-乙腈作為流動(dòng)相時(shí),諾氟沙星出峰時(shí)間為12.377 min,且基線不穩(wěn)定;0.2%甲酸-乙腈作為流動(dòng)相時(shí),諾氟沙星出峰時(shí)間為8.533 min,極大地節(jié)約了檢測(cè)時(shí)間,且響應(yīng)值較高,基線穩(wěn)定,鋒型更加對(duì)稱尖銳,分離效果較好,信噪比更高。具體見圖1、圖2。此外,在試驗(yàn)過程中發(fā)現(xiàn),熒光檢測(cè)器對(duì)于甲酸與乙腈比例的變化較為敏感,梯度洗脫時(shí)引起的基線漂移較為明顯,本文最終選擇0.2%甲酸水溶液-乙腈(87∶13)作為流動(dòng)相進(jìn)行等度洗脫,洗脫時(shí)間18 min。此條件溫和,不會(huì)損傷色譜柱,同時(shí)能夠有效分離得到較好峰形,易控制。
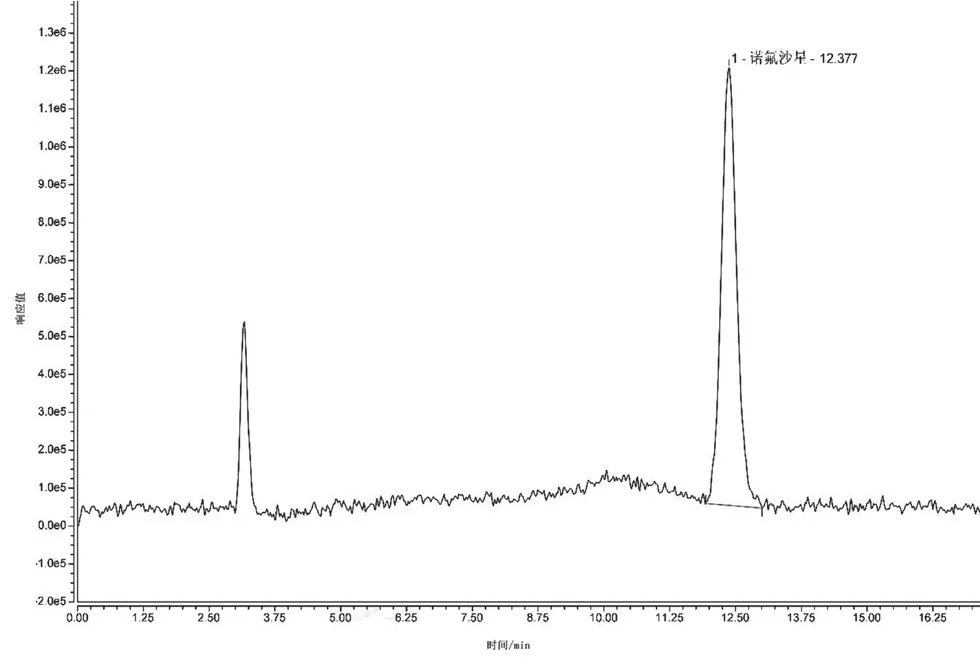
圖1 流動(dòng)相為0.05 mol·L-1磷酸鹽/三乙胺-乙腈時(shí)的色譜圖
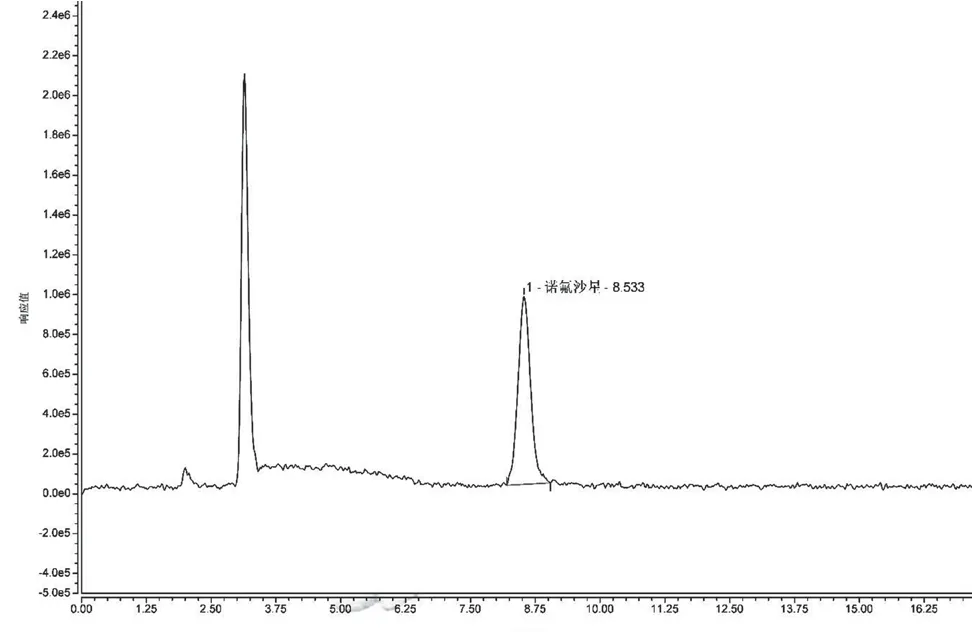
圖2 流動(dòng)相為0.2%甲酸水溶液/乙腈時(shí)的色譜圖
2.1.2 流速優(yōu)化
不同的色譜柱對(duì)同一樣品有不同的保留時(shí)間,同一物質(zhì)其出峰時(shí)間受流速的影響極大。一般情況下,流速由色譜柱的內(nèi)徑大小直接決定,內(nèi)徑越大流速則相對(duì)較大,樣品分析時(shí)間也會(huì)相對(duì)較短,所以提高流速可大大縮短樣品分析時(shí)間,但樣品在色譜柱中的流速過大則會(huì)在一定程度上影響其分離效果,引起基線漂移。本試驗(yàn)選擇 1.2 mL·min-1、1.5 mL·min-1、1.8 mL·min-13種不同流速,觀察流速對(duì)分離時(shí)間、基線和分離度的影響,結(jié)果見圖3、圖4和圖5。
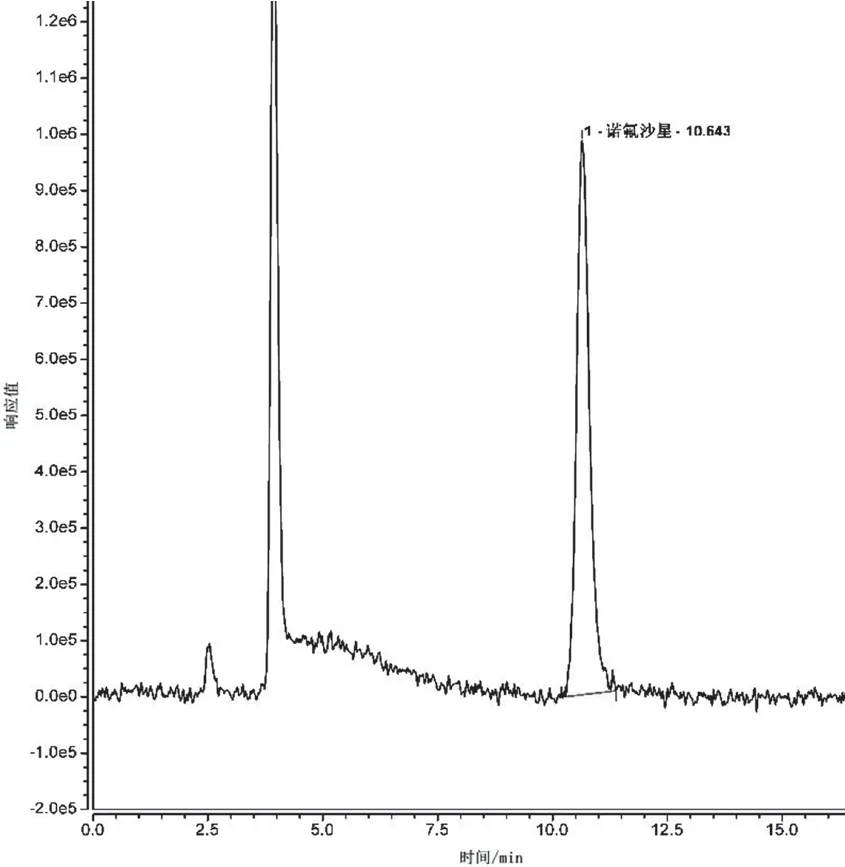
圖3 流速為1.2 mL·min-1時(shí)的色譜圖
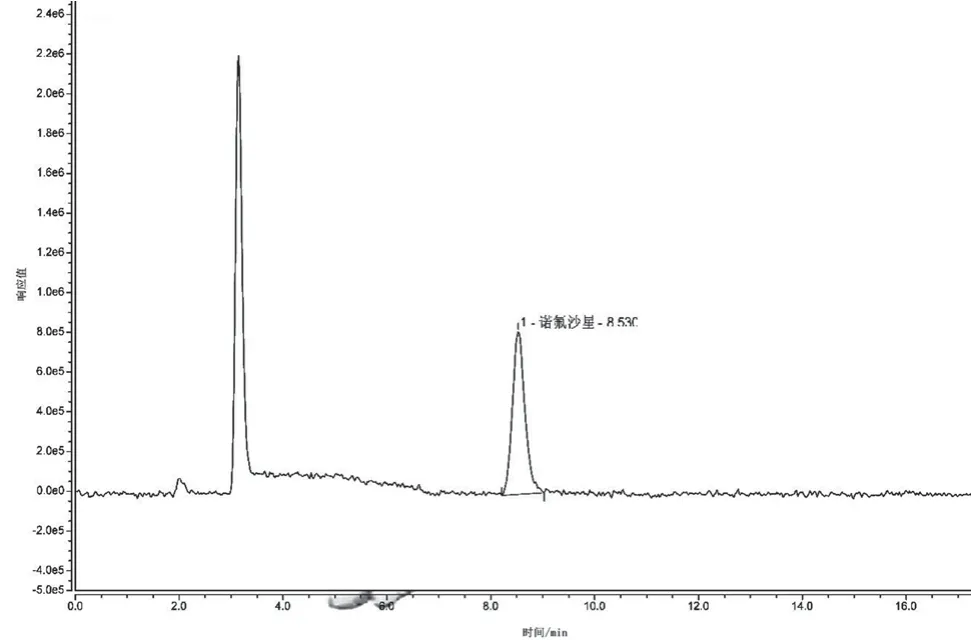
圖4 流速為1.5 mL·min-1時(shí)的色譜圖
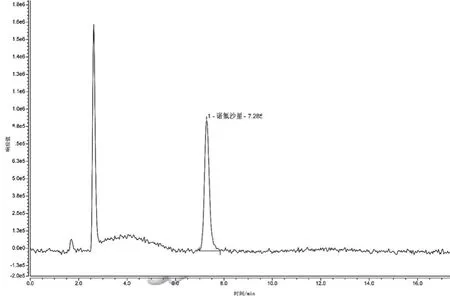
圖5 流速為1.8 mL·min-1時(shí)的色譜圖
隨著流速的增加,目標(biāo)物出峰時(shí)間逐漸縮短,流速為1.8 mL·min-1時(shí),出峰時(shí)間為7.285 min,峰圖有稍微拖尾的現(xiàn)象,出峰前基線不穩(wěn)現(xiàn)象明顯;當(dāng)流速為1.2 mL·min-1,出峰時(shí)間為10.643 min,目標(biāo)物后含有小峰;流速為1.5 mL·min-1時(shí)出峰時(shí)間為8.530 min,峰圖無拖尾現(xiàn)象且峰形較為尖銳對(duì)稱,相對(duì)流速為1.8 mL·min-1的譜圖,其基線更穩(wěn)定,峰圖效果好,相對(duì)流速為1.2 mL·min-1的譜圖,其出峰時(shí)間有所縮短,峰圖效果好,因此本試驗(yàn)研究最終選擇流速為 1.5 mL·min-1。
2.2 樣品前處理?xiàng)l件優(yōu)化
2.2.1 方法初確定
(1)稀釋液的選擇。蜂蜜是一種特殊物質(zhì),黏稠度較大,很難均勻分散,在試驗(yàn)過程中發(fā)現(xiàn)如果直接向蜂蜜中加入提取液乙腈,會(huì)使蜂蜜變成膠狀物。有研究表明,氫氧化鈉溶液有助于蜂蜜的溶解[17]。因此本試驗(yàn)選擇水、Mallvaine pH 4.0緩沖液和0.02 mol·L-1氫氧化鈉3種稀釋液,在提取之前將蜂蜜樣品制備成均勻相體系,試驗(yàn)結(jié)果發(fā)現(xiàn)等量添加水、Mallvaine pH 4.0緩沖液時(shí),蜂蜜不易溶解分散,而同比例的0.02 mol·L-1氫氧化鈉溶液可以很好地將蜂蜜溶解使其成為分散性體系,且分離程度高,色譜圖效果好,因此選擇1.3.3中方法2進(jìn)行蜂蜜稀釋液制備,即選取0.02 mol·L-1氫氧化鈉溶液作為蜂蜜樣品中稀釋液。
(2)提取液的選擇。選取0.05 mol·L-1L磷酸緩沖鹽溶液、0.5%乙酸的乙腈溶液、乙腈+水(8∶2)混合溶液作為蜂蜜中諾氟沙星的提取液。試驗(yàn)過程中發(fā)現(xiàn),同體積添加的3種提取液所提取的諾氟沙星含量分別為 0 ng·g-1、1.181 3 ng·g-1、1.151 9 ng·g-1,可見0.5%乙酸的乙腈溶液提取效果最好,因此初選取0.5%乙酸的乙腈溶液作為蜂蜜稀釋液中諾氟沙星的提取液。具體見圖6~圖9。
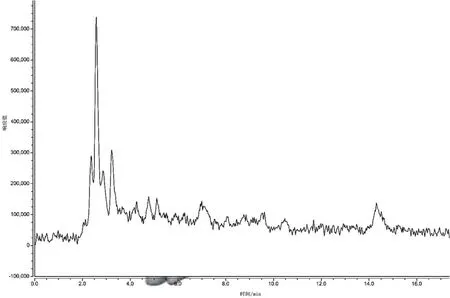
圖6 空白蜂蜜樣品色譜圖
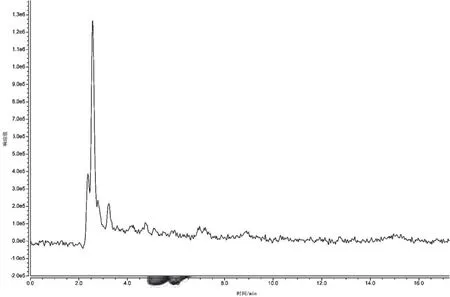
圖7 磷酸鹽緩沖液為提取液時(shí)的陽性樣品色譜圖
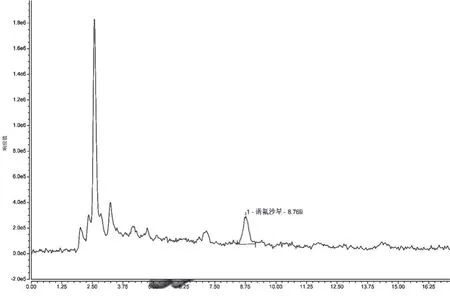
圖8 0.5%乙酸的乙腈溶液為提取液時(shí)的陽性樣品色譜圖
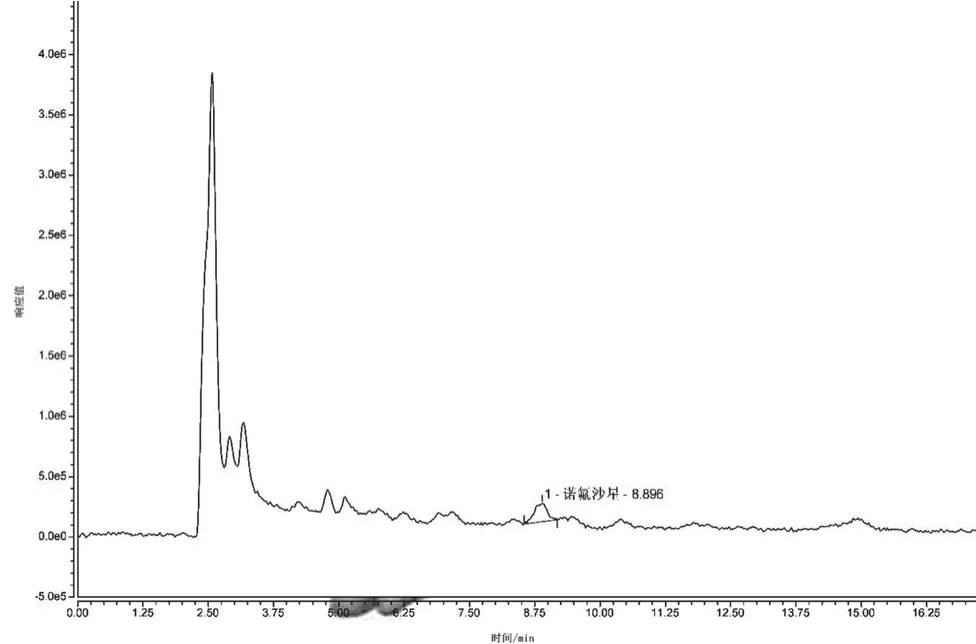
圖9 乙腈+水(8∶2)混合溶液為提取液時(shí)的陽性樣品色譜圖
2.2.2 方法的進(jìn)一步確定與優(yōu)化
在大量的預(yù)試驗(yàn)基礎(chǔ)上發(fā)現(xiàn),樣品稀釋液的添加量和提取時(shí)加入0.5%乙酸的乙腈溶液的量對(duì)諾氟沙星的提取濃度有明顯影響,本文利用2因素3水平試驗(yàn),研究這2個(gè)因素對(duì)試驗(yàn)結(jié)果的影響,結(jié)果分析如表1所示。
由表1可知,當(dāng)蜂蜜∶0.02 mol·L-1氫氧化鈉溶液=1∶1,蜂蜜∶0.5%乙酸的乙腈溶液=1∶5時(shí),諾氟沙星提取效果最佳,提取量為1.349 6 ng·g-1。
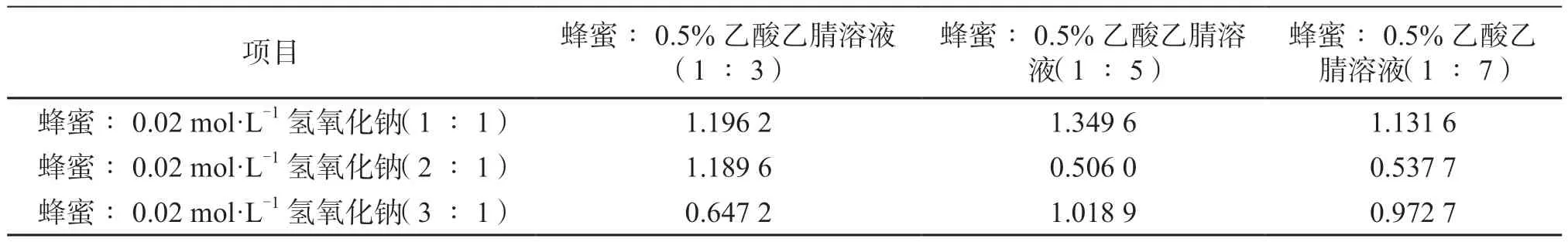
表1 不同添加量的氫氧化鈉和乙酸乙腈溶液對(duì)諾氟沙星提取濃度的影響(單位:ng·g-1)
2.2.3 重復(fù)性驗(yàn)證
當(dāng)蜂蜜∶0.02 mol·L-1氫氧化鈉溶液=1∶1,蜂蜜∶0.5%乙酸的乙腈溶液=1∶5時(shí),添加不同濃度諾氟沙星,驗(yàn)證蜂蜜中諾氟沙星的重復(fù)性驗(yàn)證,結(jié)果分析如表2所示。選擇含3.00 ng·mL-1、8.70 ng·mL-1諾氟沙星的蜂蜜樣品,經(jīng)測(cè)試精密度變異系數(shù)均不超過10%。
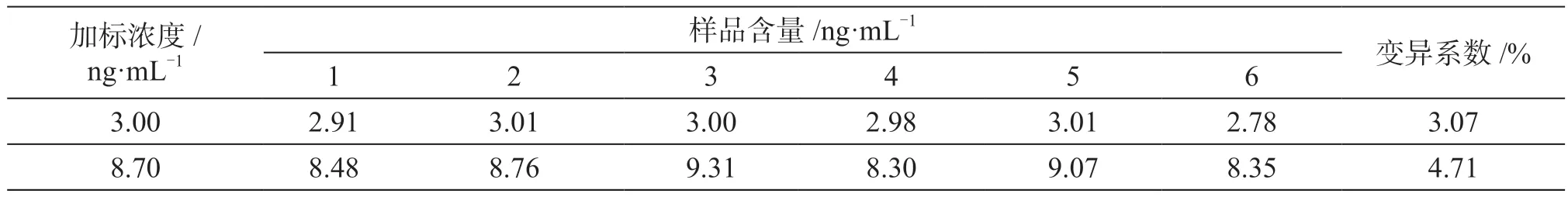
表2 不同添加量的諾氟沙星重復(fù)性驗(yàn)證結(jié)果
2.3 方法驗(yàn)證
2.3.1 標(biāo)準(zhǔn)曲線及線性范圍
將諾氟沙星標(biāo)準(zhǔn)液逐步稀釋,配制成濃度為0.001 μg·mL-1、0.002 μg·mL-1、0.004 μg·mL-1、0.010 μg·mL-1和 0.200 μg·mL-1標(biāo) 準(zhǔn) 工 作 液, 以 目標(biāo)峰面積為縱坐標(biāo)(Y)對(duì)進(jìn)樣濃度(X)進(jìn)行線性擬合,得到的標(biāo)準(zhǔn)曲線方程為Y=162 611 326.87X-422 884.11,在 0.001 ~ 0.200 μg·mL-1線性關(guān)系良好,相關(guān)系數(shù)R2為0.995 7。
2.3.2 精密度與檢出限
精密吸取標(biāo)液0.003 μg·mL-1,平行測(cè)定6次,得到各組分相對(duì)標(biāo)準(zhǔn)偏差為0.72%。在空白蜂蜜基質(zhì)中添加0.4 μg·kg-1諾氟沙星標(biāo)準(zhǔn)溶液,諾氟沙星藥物的信噪比(S/N)大于10,表明該方法諾氟沙星藥物的方法檢出限可達(dá)到0.4 μg·kg-1,滿足分析要求。
2.3.3 加標(biāo)試驗(yàn)
向5 g蜂蜜空白樣中添加濃度分別為0.03 μg·mL-1、0.05 μg·mL-1、0.10 μg·mL-1的 諾 氟 沙 星 標(biāo) 液 各100 μL進(jìn)行加標(biāo)回收試驗(yàn),每個(gè)加標(biāo)水平重復(fù)3次。諾氟沙星回收率結(jié)果見表3。其回收率為84.98%~105.72%,相對(duì)標(biāo)準(zhǔn)偏差為1.84%~2.94%,滿足線性分析要求。
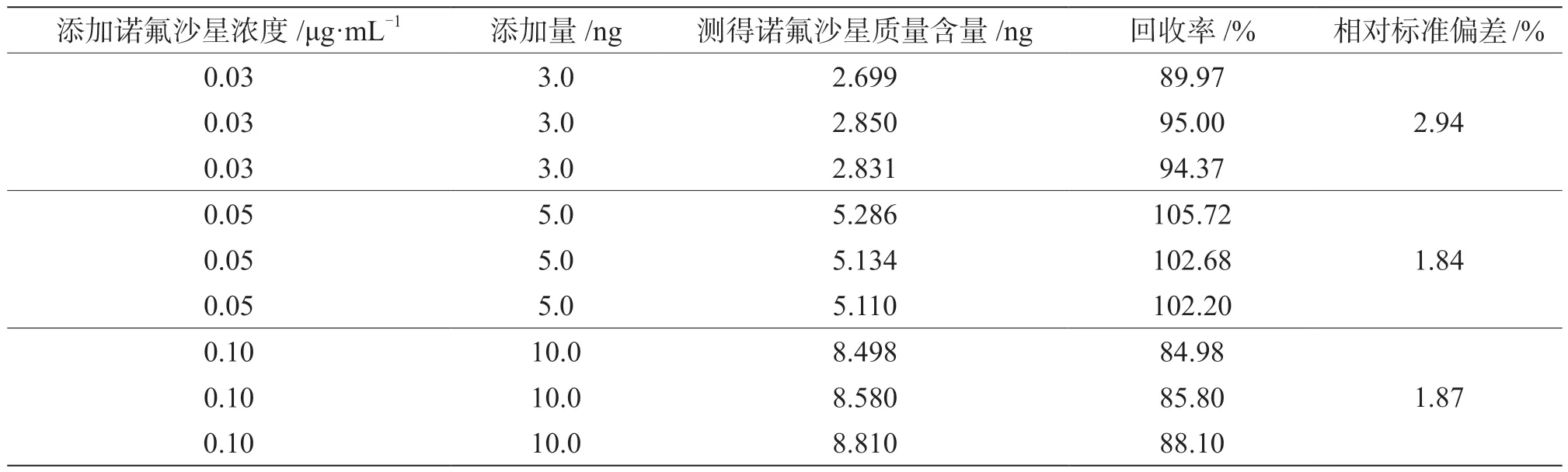
表3 蜂蜜中添加不同濃度標(biāo)樣所測(cè)回收率
2.3.4 蜂蜜產(chǎn)品檢測(cè)
為了將方法應(yīng)用于市場(chǎng),對(duì)市場(chǎng)上的蜂蜜樣品任選3個(gè)種類、5種不同品牌進(jìn)行檢測(cè),分別為椴樹蜂蜜、洋槐蜂蜜及老蜂蜜共抽檢了5份,均未檢出諾氟沙星,符合我國規(guī)定蜂蜜中諾氟沙星的標(biāo)準(zhǔn)值(不得檢出),差異不顯著。
3 結(jié)論
本文對(duì)市場(chǎng)上的蜂蜜樣品任選3個(gè)種類、5種不同品牌進(jìn)行檢測(cè),均未檢出諾氟沙星,符合我國規(guī)定蜂蜜中諾氟沙星的標(biāo)準(zhǔn)值(不得檢出),差異不顯著。以乙腈-2%甲酸水溶液(13∶87)為流動(dòng)相,流速為1.5 min·L-1,柱溫35 ℃,熒光檢測(cè)激發(fā)波長(zhǎng)280 nm,發(fā)射波長(zhǎng)450 nm為優(yōu)化后的色譜條件;以0.02 mol·L-1氫氧化鈉溶液(與蜂蜜的比例為1∶1)溶解蜂蜜,制作成蜂蜜溶液,然后用0.5%乙酸的乙腈溶液與蜂蜜水溶液(5∶1)萃取諾氟沙星,過固相萃取柱氮?dú)獯蹈桑僖砸译嫒芙鉃閮?yōu)化后的前處理方法,采用高效液相色譜法-熒光檢測(cè)器檢測(cè)蜂蜜中的諾氟沙星藥物殘留量。結(jié)果表明,該方法的檢出 限 為 0.4 μg·kg-1, 在 0.001 ~ 0.200 μg·mL-1呈 良好的線性關(guān)系,相關(guān)系數(shù)R2=0.995 7,加標(biāo)回收率為84.98%~105.72%,利用該方法通過檢測(cè)不同品牌生產(chǎn)的蜂蜜,結(jié)果均未檢出諾氟沙星,進(jìn)一步驗(yàn)證本方法能夠滿足殘留分析的要求,為蜂蜜中諾氟沙星殘留量測(cè)定提供了一種簡(jiǎn)便快捷準(zhǔn)確的色譜方法,為蜂產(chǎn)業(yè)的安全生產(chǎn)提供了保障。

- 食品安全導(dǎo)刊的其它文章
- 植物基食品的規(guī)范標(biāo)準(zhǔn)適用問題和消費(fèi)認(rèn)知挑戰(zhàn)
- 安徽省推行網(wǎng)絡(luò)餐飲食安封簽工作推進(jìn)會(huì)在蕪湖市召開
- 河南省鄭州市市場(chǎng)監(jiān)督管理局對(duì)高等院校和醫(yī)療機(jī)構(gòu)食品安全開展專項(xiàng)檢查
- 采用更精準(zhǔn)、非靶向鑒別手段,保證食品真實(shí)性
——探索高通量基因測(cè)序技術(shù)未來發(fā)展之路 - 月餅盒包裝設(shè)計(jì)的優(yōu)化和創(chuàng)新方法研究
- 益生菌微膠囊技術(shù)對(duì)益生菌存活率影響的研究進(jìn)展