右美托咪定通過調控Nrf2減輕HT22細胞缺氧/復氧引起的鐵死亡
2022-10-14 11:53:32門運政段方方童旭輝金文靜董淑英
中國藥理學通報 2022年10期
門運政,胡 淼,劉 剛,段方方,童旭輝,黃 杰,金文靜,董淑英,3
(蚌埠醫學院 1.藥學院藥理學教研室、2.第一附屬醫院麻醉科、3.心腦血管疾病基礎與臨床重點實驗室,安徽 蚌埠 233030)
腦卒中是嚴重危害人類健康的疾病,其中缺血性腦卒中占比達80%以上。目前臨床上主要采用溶栓和血管內介入療法以恢復血管再通[1],然而血液恢復灌注時又會造成腦缺血/再灌注(ischemia/reperfusion,I/R)損傷。
鐵死亡是一種鐵依賴的脂質過氧化物的過量累積導致的細胞死亡。大量臨床數據表明,腦缺血患者腦組織中的鐵水平明顯升高。另有研究報道,香芹酚通過增加谷胱甘肽過氧化物酶4(glutathione peroxidase 4,GPX4)的表達,抑制沙鼠腦I/R損傷中的鐵死亡,進而提高沙鼠腦I/R損傷中海馬神經元的存活率[2],但鐵死亡在腦I/R損傷中具體的作用機制,仍有待進一步的研究。
核因子E2相關因子2(nuclear factor erythroid 2-related factor 2,Nrf2)是胞內的重要轉錄因子,能夠協調胞內各種抗氧化基因在應激狀態下的激活,可通過影響脂質過氧化的生成進而調控鐵死亡。有研究表明,Nrf2可以通過調節其下游基因的轉錄進而提高腫瘤細胞對鐵死亡的抵抗能力[3]。胱氨酸/谷氨酸逆向轉運蛋白溶質載體家族7成員11(solute carrier family 7 member 11,SLC7A11)是編碼谷胱甘肽合成相關蛋白的基因之一,作為Nrf2的下游,具有抵抗脂質過氧化的能力。有研究表明,SLC7A11表達被抑制后,可導致GPX4活性和抵抗脂質過氧化的能力降低,最終發生鐵死亡[4]。然而,Nrf2是否參與了腦I/R損傷中鐵死亡的發生仍不清楚。
右美托咪定(dexmedetomidine,DEX)是一種臨床上廣泛使用的麻醉輔助用藥,具有鎮靜、抗焦慮和鎮痛作用[5]。研究表明,DEX對多種組織器官的I/R損傷均具有保護作用[6],例如心臟、腎臟和腦等。有文獻報道,在SK-N-SH細胞中,DEX的抗氧化作用與其抑制鐵超載有關[7]。大量研究表明,細胞內鐵離子的超載是誘導細胞鐵死亡的主要機制[8]。然而,DEX在腦I/R損傷中的保護作用,是否與其對鐵死亡的抑制有關,卻尚未見報道。因此本研究采用HT22細胞H/R損傷模型,探討DEX抗HT22細胞H/R損傷的作用及其機制,為DEX在臨床治療腦I/R損傷提供理論依據。
1 材料與方法
1.1 細胞HT22細胞購買于中科院上海細胞庫。
1.2 主要試劑鹽酸右美托咪定注射液(貨號H20110076)購自江蘇恩華藥業有限公司;DMEM干粉(貨號2164119)購自美國Gibco公司;ML385(貨號HY-100523)購自美國MCE公司,丙二醛試劑盒(貨號A003-1)、還原型谷胱甘肽試劑盒(貨號A006-2-1)購自南京建成生物工程研究所,胰蛋白酶(貨號EZ2811A172)購自中國上海Beyotime公司;Nrf2抗體(貨號ab137550)、GPX4抗體(貨號ab125066)、SLC7A11抗體(貨號ab37185)、TFR1抗體(貨號ab8403 )購自英國Abcam公司;GAPDH抗體(貨號10494-1-AP)購自中國Proteintech公司;FerroOrange熒光探針(貨號SH951)購自日本DOJINDO公司,C11 BODIPY 581/591(貨號2115343)購自美國Sigma公司。
1.3 主要儀器酶標儀購自美國BioiTek公司,高速冷凍離心機購自德國艾本德公司,凝膠成像儀購自美國BIO-RAD公司,二氧化碳培養箱購自上海ESCO公司,缺氧培養箱購自美國賽默飛公司。
1.4 H/R損傷模型建立當HT22細胞占據培養皿65%左右時,用PBS清洗細胞3次,用無血清無糖培養基替換完全培養基至細胞培養皿中,置于缺氧培養箱(94% N2,5% CO2,1% O2),缺氧12 h,復糖復氧24 h。
1.5 實驗分組HT22細胞分為:,對照組(Control)、H/R組、DEX低濃度組(H/R+DEX2.5,μmol·L-1)組、DEX中濃度組(H/R+DEX5,5 μmol·L-1)組、DEX高濃度組(H/R+DEX10,10 μmol·L-1)組,探討DEX對HT22細胞H/R損傷的保護作用的最適濃度;再將HT22細胞分為4組,即Control、H/R組、H/R+DEX5組和H/R+DEX5+ML385組,進一步探究DEX對HT22細胞H/R保護作用的機制。DEX在缺氧時加入;ML385于復氧時立即加入,終濃度為2 μmol·L-1。
1.6 細胞存活率檢測HT22細胞接種于96孔板中,分組給藥并處理后,每孔加入20 μL MTT試劑,繼續孵育4 h。孵育完成后,于孔中加入DMSO,37 ℃烘箱中孵育0.5 h。孵育完成后檢測各孔在490 nm波長處的吸光度值,并計算細胞存活率。
1.7 細胞中Fe2+的檢測消化并收集細胞,將HT22細胞接種于共聚焦小皿中,待細胞長至適宜密度時,根據分組,給藥處理。處理后棄去培養液,加入FerroOrange工作液,37 ℃孵育20 min。活細胞工作站下觀察細胞,并進行圖像采集。
1.8 細胞內Lipid ROS檢測取對數生長期的細胞,接種于6孔板中培養,按照實驗分組完成相應的處理,加入C11 BODIPY 581/591染液,在培養箱中孵育0.5 h。棄去各孔中的培養液,消化并收集細胞,隨后用250 μL PBS重懸,最后將重懸液通過流式細胞儀檢測,并數據收集。
1.9 MDA、GSH含量檢測不同實驗組的細胞,給藥處理后,消化并收集細胞,超聲破碎后按照MDA,GSH說明書中的操作于532 nm、405 nm處檢測各實驗組細胞中MDA、GSH的含量。
1.10 Western blot檢測蛋白收集并裂解細胞,隨后進行定量、上樣、轉膜、封閉,孵育一抗過夜(Nrf2、TFR1、SLC7A11、GPX4抗體稀釋比例均為1 ∶1 500,GAPDH抗體稀釋比例為1 ∶3 000)。次日孵育二抗,隨后用凝膠成像儀對蛋白條進行成像并用Bio Imaging System對圖像進行灰度掃描分析。
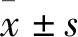
2 結果
2.1 DEX對H/R損傷HT22 細胞存活率的影響結果顯示,經H/R處理后HT22細胞的存活率明顯降低(P<0.01);H/R+DEX2.5、H/R+DEX5、H/R+DEX10組HT22細胞存活率相較于H/R組均有不同程度的升高(P<0.05),且H/R+DEX5組與H/R+DEX10組HT22細胞存活率升高較為明顯(P<0.01)。同時H/R+DEX5,H/R+DEX10兩組細胞存活率無明顯差異,所以后續均采用5 μmol·L-1的DEX開展實驗。B圖顯示,在使用DEX的基礎上使用Nrf2抑制劑ML385(2 μmol·L-1)[9]后,HT22細胞存活率較H/R+DEX5組降低(P<0.01)。
2.2 DEX對H/R損傷HT22細胞中Nrf2 蛋白表達的影響結果顯示,相較于Control組,H/R損傷后的Nrf2蛋白表達水平明顯降低(P<0.01);相較于H/R組,DEX處理后中Nrf2蛋白表達明顯增加(P<0.05);相較于H/R+DEX組,在DEX基礎上使用ML385后,Nrf2蛋白表達又明顯降低(P<0.01)。
2.3 DEX對H/R損傷HT22細胞中Fe2+、Lipid ROS、MDA及GSH的影響采用FerroOrange作為熒光探針,對細胞內Fe2+進行熒光成像。結果顯示,與Control組相比較,H/R組Fe2+熒光強度增強,表明細胞中的Fe2+含量增加;與H/R組相比,H/R+DEX5組Fe2+熒光強度減弱,細胞內Fe2+含量減少;與H/R+DEX5組相比,H/R+DEX5+ML385組Fe2+熒光強度增強,胞內Fe2+含量增加。

Fig 1 Effect of DEX and ML385 on survival rates of HT22 cells with H/R n=3)A:Different concentrations of DEX increased the survival fraction of HT22 cells with H/R injury;B:ML385 attenuated the protective effect of DEX with H/R injury on HT22 cells n=3).##P<0.01 vs Control;△P<0.05,△△P<0.01 vs H/R;**P<0.01 vs H/R+DEX5+ML385.

Fig 2 Effect of DEX on expression of Nrf2 protein on HT22 cells with H/R injury n=3)##P<0.01 vs Control;△P<0.05 vs H/R;**P<0.01 vs H/R+DEX5+ML385
用流式細胞儀分析各實驗組細胞中Lipid ROS含量以及對胞內MDA和GSH的含量測定,結果表明:相較于Control組,H/R組HT22細胞內Lipid ROS及MDA的含量明顯升高(均P<0.01)、GSH的含量則明顯降低(P<0.01);使用DEX后,與H/R組相比,胞內Lipid ROS及MDA含量明顯降低(均P<0.01),GSH 的含量則明顯增加(P<0.01);而在使用DEX的基礎上使用Nrf2抑制劑后,胞內Lipid ROS水平及MDA含量明顯升高(均P<0.05),GSH的含量則明顯降低(P<0.01)。
2.4 DEX對H/R損傷HT22細胞中GPX4、SLC7A11和TFR1蛋白表達的影響采用蛋白質印跡法檢測HT22細胞中鐵死亡相關蛋白:GPX4、SLC7A11以及TFR1蛋白的表達變化。結果如圖所示,HT22細胞經H/R處理后,細胞中GPX4、SLC7A11蛋白表達明顯降低(均P<0.05),TFR1蛋白表達則明顯升高(P<0.01);在給予DEX后發現,GPX4、SLC7A11蛋白的表達明顯增加(均P<0.05),而TFR1蛋白的表達明顯降低(P<0.01);在DEX的基礎上使用Nrf2抑制劑后,HT22細胞中GPX4、SLC7A11蛋白表達明顯降低(均P<0.01),TFR1蛋白表達則明顯升高(P<0.01)。

Fig 3 Effect of DEX on Fe2+,lipid ROS,MDA and GSH on HT22 cells with H/R n=3)A:The fluorescence image of Fe2+ in different groups;B,C:The levels of Lipid ROS in HT22 cells with different treatment;D:MDA levels on HT22 cells with different treatment.E:GSH levels on HT22 cells with different treatment.##P<0.01 vs Control;△△P<0.01 vs H/R;*P<0.05,**P<0.01 vs H/R+DEX5+ML385

Fig 4 Effect of DEX on expression of ferroptosis related proteins on HT22 cells with H/R injury n=3)#P<0.05,##P<0.01 vs Control;△△P<0.01 vs H/R;**P<0.01 vs H/R+DEX5+ML385
3 討論
近年來,腦卒中在中老年群體中的發病率逐年增加,給患者及其家屬帶來了嚴重的負擔。DEX作為臨床常用的麻醉輔助劑,具有良好的鎮靜、抗焦慮等作用。有研究發現,DEX可通過AKT信號通路減輕大鼠心肌I/R損傷[10],Yin等[11]研究發現,DEX和Netrin-1聯合使用可以通過調節ERK5/MEF2A信號通路抑制內質網應激減輕腦I/R損傷。目前關于DEX抗腦I/R損傷的研究愈發深入,但其可能的保護機制尚需進一步探明。為了探究DEX對HT22細胞H/R損傷后的保護作用,本實驗采用MTT法檢測不同濃度的DEX對HT22細胞H/R后的生存情況。結果顯示:低、中、高濃度的DEX均能增加HT22細胞在H/R后的存活率,說明DEX能夠減輕HT22細胞H/R損傷。與單獨使用DEX相比較,在DEX基礎上使用Nrf2抑制劑ML385后,細胞存活率明顯降低,這表明Nrf2可能參與了DEX對HT22細胞H/R損傷的保護作用。
鐵死亡涉及到胞內鐵穩態的失衡和脂質過氧化的累積,是誘導腦I/R損傷的機制之一。在神經系統中,鐵穩態可維持胞內脂質、蛋白質、碳水化合物和DNA的氧化和修飾[2]。然而,鐵離子在應激條件下會在細胞內過度累積,過多的Fe2+通過芬頓(Fenton)反應引起膜脂質過氧化,導致Lipid ROS過度累積,線粒體結構皺縮,功能損傷,最終造成細胞死亡[12]。流行病學研究已經確定了鐵蓄積會加重腦卒中患者的神經損傷[13]。轉鐵蛋白受體1(transferrin receptor 1,TFR1)是胞內鐵代謝的重要蛋白,腦I/R損傷時,TFR1的表達增強,進而增加其對鐵的攝取,最終導致胞內的鐵離子過載[14]。我們的研究發現DEX可以明顯抑制HT22細胞H/R后TFR1的表達,同時,FerroOrange熒光探針的結果也表明使用DEX后可以明顯降低胞內Fe2+的水平。上述結果表明,DEX可能通過抑制TFR1的表達,進而降低由H/R引起的胞內鐵離子的過量蓄積。
正常細胞中,膜脂質過氧化反應的產物被胞內的抗氧化體系所清除,其中以SLC7A11/GPX4為核心的抗脂質過氧化體系可以將脂質過氧化物的過氧鍵轉變為羥基,失去其過氧化物的活性[15]。神經細胞H/R損傷后,伴隨著抗脂質過氧化體系的破壞,SLC7A11、GPX4蛋白的表達降低,以及胞內抗氧化物GSH含量的大幅減少。因此鐵蓄積和抗脂質過氧化體系的損傷導致了大量Lipid ROS的產生以及生物膜脂質過氧化產物MDA的累積。而DEX具有優秀的抗氧化能力,可以通過清除細胞內過多的氧自由基減輕氧化損傷[16]。在HT22細胞H/R損傷中,使用DEX后明顯提高SLC7A11和GPX4的蛋白表達以及胞內GSH的水平,DEX對于HT22細胞抗氧化體系的保護顯然增強了其對脂質過氧化物的分解能力:Lipid ROS和MDA的水平相較于H/R損傷均明顯下降。
Nrf2是具有神經保護作用的關鍵轉錄因子,負責維持細胞氧化還原的動態平衡。研究表明,人參皂苷Rg1可以通過激活Nrf2減輕心肌細胞H/R損傷[17]。此外,在腦I/R損傷中的研究表明,激活Nrf2可以通過抑制神經炎癥和氧化應激減輕神經損傷[18]。我們的研究發現,DEX可以明顯提高H/R損傷下HT22細胞中Nrf2的表達,為了進一步探究Nrf2在DEX抗HT22細胞H/R損傷中的作用,我們使用ML385抑制Nrf2的表達,實驗結果表明,Nrf2被抑制后DEX對胞內亞鐵離子的運輸能力明顯降低,同時,胞內的各種抵抗脂質過氧化的能力及脂質過氧化產物均明顯下降,這表明Nrf2的抑制降低了DEX對細胞H/R損傷引起的鐵死亡的抵抗能力。
鐵在氧氣運輸,能量代謝和DNA合成中均發揮著重要的作用,然而過量的鐵對于細胞內氧化還原穩態是有害的。同樣,鐵離子依賴的胞內脂質氧化穩態的失衡是導致神經細胞H/R損傷的重要原因。我們的研究表明DEX可以通過Nrf2抑制TFR1的表達,調控受損狀態下神經細胞鐵離子的運輸,進而維持胞內鐵離子水平的穩態。同時,鐵離子作為芬頓反應的重要參與者,當其在胞內過度蓄積的情況得以緩解時,芬頓反應也會受到抑制。此外,DEX經由Nrf2增強了神經細胞以SLC7A11/GPX4為核心的抗脂質過氧化的能力,最終使得脂質過氧化產物Lipid ROS以及MDA水平的降低。本研究以鐵死亡為切入點,探究了DEX在抗HT22細胞H/R損傷中的作用及機制,為臨床上合理使用DEX提供了新的理論依據。
