3-異硫氰酸酯氧化吲哚在串聯環化反應中的研究進展
2022-10-14 01:45:08張顏萍王振華趙建強尹俊青袁偉成
成都大學學報(自然科學版) 2022年3期
關鍵詞:催化劑
張顏萍,游 勇,王振華,趙建強,尹俊青,袁偉成
(1.成都大學 高等研究院,四川 成都 610106; 2.成都大學 藥學院,四川 成都 610106)
0 引 言
立體選擇性構建手性雜環化合物是現代有機合成的重要研究方向,同時,手性單環和多環體系在天然產物和藥物中的重要作用也推動了該領域的發展[1-5].噁唑烷-2-(硫)酮[6-14]、咪唑烷-2-(硫)酮[15-16]與吡咯烷-2-(硫)酮[17-21]衍生物是有機合成和藥物化學中的重要結構單元.其中,具有光學活性的噁唑烷-2-(硫)酮衍生物可用于生物活性和藥物活性的化合物及天然產物的全合成.與環硫代氨基甲酸酯相比,噁唑烷-2-(硫)酮衍生物可以很容易地轉化為β-羥基α-氨基化合物[6-11].近年來,α-異硫氰酸酯類化合物參與的不對稱[3+2]環化反應構建噁唑烷-2-(硫)酮、咪唑烷-2-(硫)酮與吡咯烷-2-(硫)酮等衍生物受到化學工作者的廣泛關注.目前報道的α-異硫氰酸酯類化合物主要有α-異硫氰酸酯酯、α-異硫酸酯酰胺與α-異硫氰酸酯磷酸酯3種類型.其中,本課題組首次報道了3-異硫氰酸酯氧化吲哚的合成及其參與的不對稱反應[22].通過3-異硫氰酸酯氧化吲哚參與的串聯環化反應,可以高效構建一系列含有噁唑烷-2-(硫)酮、咪唑烷-2-(硫)酮與吡咯烷-2-(硫)酮結構的螺環氧化吲哚骨架,該類骨架是許多具有生物活性和藥理活性的化合物的關鍵結構單元(見圖1).本文總結了本課題組在有機小分子催化和金屬催化3-異硫氰酸酯氧化吲哚參與的Aldol/cyclization反應、Mannich/cyclization反應、Michael/cyclization反應、不對稱去芳構化[3+2]環化反應與[3+3]環化反應方面的研究工作.
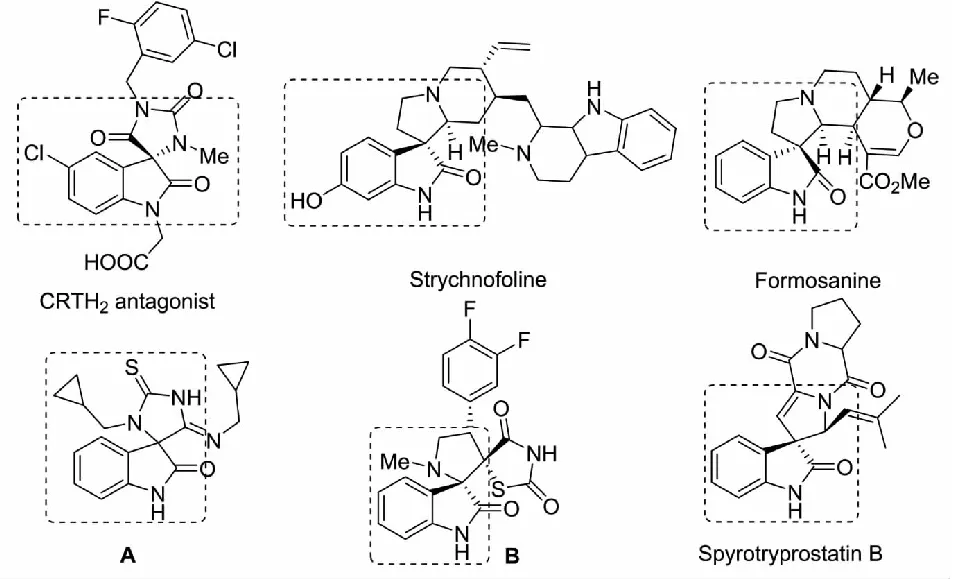
圖1 具有生物活性的螺環氧化吲哚骨架結構化合物
1 Aldol/cyclization反應
3-異硫氰酸酯氧化吲哚與醛、酮的Aldol/cyclization反應是構建噁唑烷-2-硫酮結構單元的高效方法之一[23-26],并且,由于螺環氧化吲哚結構單元廣泛存在于臨床藥物和天然生物堿中,因此,噁唑烷-2-硫酮螺氧化吲哚衍生物成為有機合成和藥物化學研究者重點關注的化合物.
2011年,本課題組首次設計并合成了一類新型的α-異硫氰酸酯酰胺,即3-異硫氰酸酯氧化吲哚1.并報道了在1,2-二苯基乙二胺衍生的手性雙功能催化劑3的作用下,3-異硫氰酸酯氧化吲哚1與酮2的不對稱Aldol/cyclization反應,以高達95%的收率,95∶5的非對映選擇性(dr值)和98%的對映選擇性(ee值)構建了一系列噁唑烷-2-硫酮螺氧化吲哚化合物4,這是首例3-異硫氰酸酯氧化吲哚參與不對稱反應得到螺環氧化吲哚的報道(見圖2)[22].通過這一方法可以有效合成一系列復雜結構的螺環氧化吲哚衍生物.該反應對各類底物具有良好的普適性,采用不同取代的苯乙酮、β-萘乙酮與環己酮,反應均可順利進行.3-異硫氰酸酯氧化吲哚苯環上取代基的電子性質對反應的活性和立體選擇性的影響不大(4d-4e),將3-異硫氰酸酯氧化吲哚的N-Me替換成N-Bn時,仍以92%的收率,90∶10的dr值和90%的ee值得到了目標化合物.
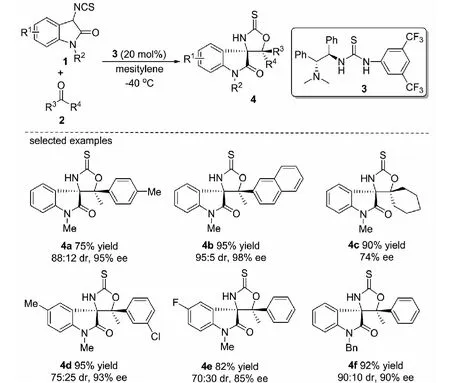
圖2 3-異硫氰酸酯氧化吲哚與酮的不對稱Aldol/cyclization反應
受到這一工作的啟發,2013年,課題組又報道了3-異硫氰酸酯氧化吲哚1與醛5的不對稱Aldol/cyclization反應,在1,2-二苯基乙二胺-硫脲3的催化作用下,以高達95%的收率,98∶2的dr值和89%的ee值得到一系列含有相鄰季碳-叔碳手性中心的噁唑烷-2-硫酮螺氧化吲哚衍生物6(見圖3)[27].在考察底物普適性時發現,帶有不同取代基的3-異硫氰酸酯氧化吲哚大多數具有較高的反應活性,與醛反應1 min就可結束.
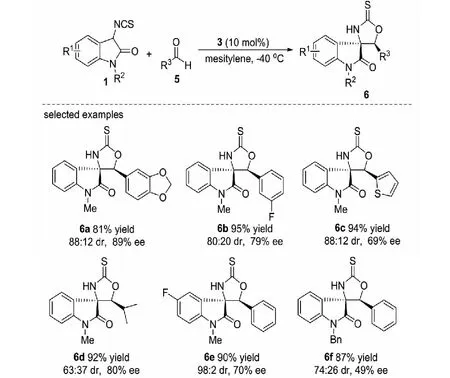
圖3 3-異硫氰酸酯氧化吲哚與醛的不對稱Aldol/cyclization反應
該課題組同時提出了反應可能的過渡態.1,2-二苯基乙二胺-硫脲3的叔胺部分奪取3-異硫氰酸酯氧化吲哚3位的質子,并與其烯醇互變異構體形成分子內的氫鍵;與此同時,催化劑硫脲部分的雙N-H鍵通過與醛的羰基氧形成雙氫鍵來活化醛.在此過渡態下,活化的3-異硫氰酸酯氧化吲哚更易進攻醛發生Aldol反應;接著,氧負離子進攻3-異硫氰酸酯氧化吲哚NCS基團中的碳原子而發生分子內的環化反應,最終生成螺環氧化吲哚產物(見圖4).這一過渡態充分體現了1,2-二苯基乙二胺-硫脲作為手性催化劑在活化親核試劑和親電試劑的雙功能特性.
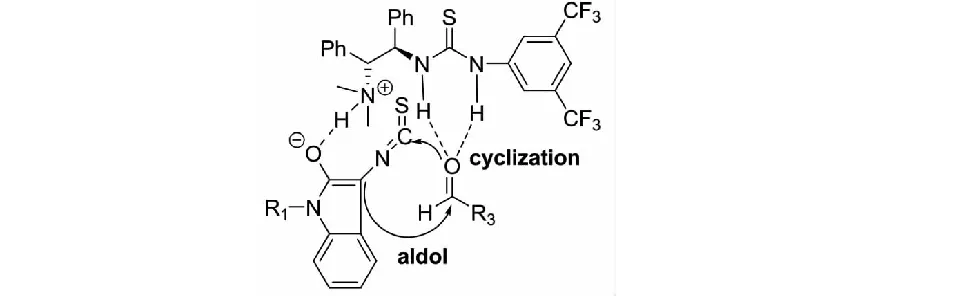
圖4 3-異硫氰酸酯氧化吲哚與醛反應的可能過渡態
2012年,課題組報道了3-異硫氰酸酯氧化吲哚1與靛紅7的Aldol/cyclization反應,以1 mol%的三乙胺為催化劑,能以高達97%的收率和99∶1的dr值得到一系列螺環氧化吲哚的衍生物8(見圖5)[28].
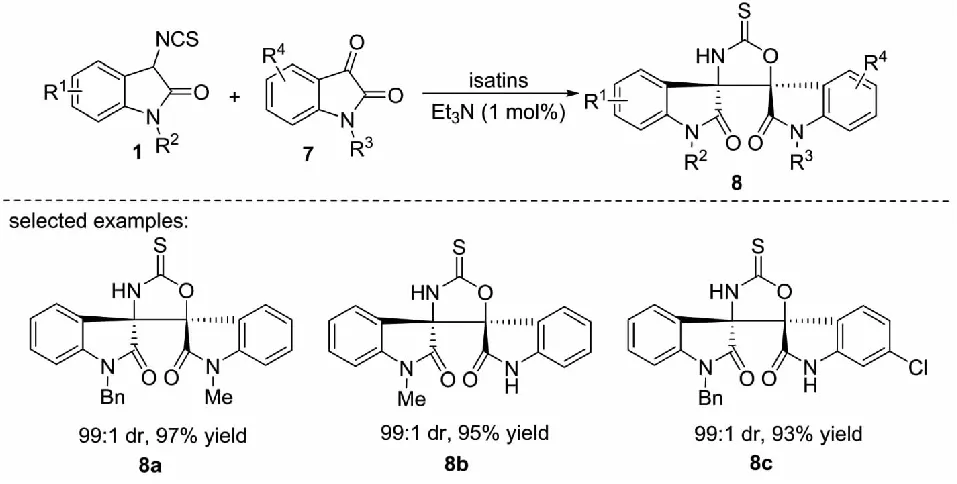
圖5 3-異硫氰酸酯氧化吲哚與靛紅的Aldol/cyclization反應
2 Mannich/cyclization反應
α-異硫氰酸酯類化合物與亞胺的Mannich/cyclization串聯反應是構建咪唑烷-2-硫酮衍生物的高效方法之一.基于叔胺-硫脲催化的3-異硫氰酸酯氧化吲哚在不對稱Aldol/cyclization反應中取得顯著成功的例子,2012年,課題組報道了3-異硫氰酸酯氧化吲哚1與靛紅亞胺9的Mannich/cyclization反應,以1 mol%的三乙胺為催化劑,能以高達97%的收率和99∶1的dr值得到一系列螺環氧化吲哚的衍生物10(見圖6)[28].此反應的優點是反應條件溫和,產物具有雙螺環氧化吲哚的骨架結構.此外,該非對映體產物可以很容易通過柱層析分離.
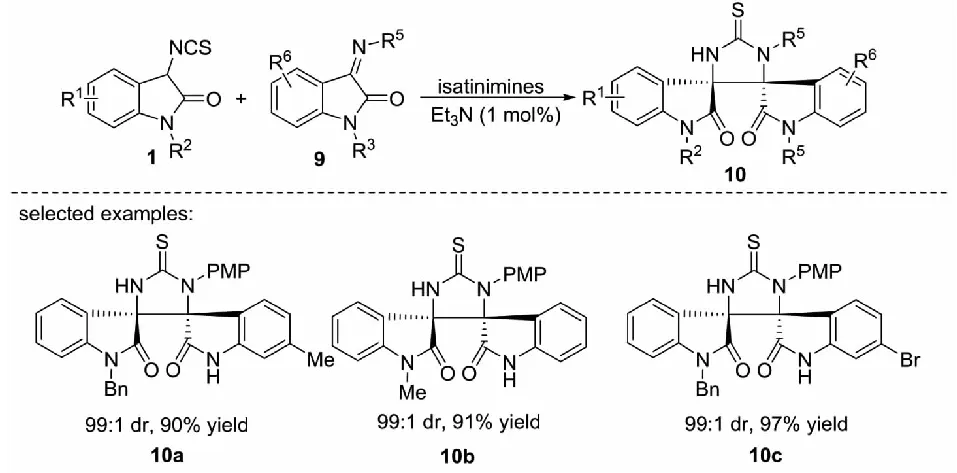
圖6 3-異硫氰酸酯氧化吲哚與靛紅亞胺的串聯反應
2015年,課題組報道了以商業化的奎寧12為催化劑,3-異硫氰酸酯氧化吲哚與對甲苯磺酰基(Ts)保護的醛亞胺11發生不對稱Mannich/cyclization反應,以高達99%的收率,大于99∶1的dr值和97%的ee值構建一系列咪唑烷-2-硫酮螺氧化吲哚衍生物13(見圖7)[29].另外,該催化體系對其他類型的亞胺也具有良好的底物普適性,例如,N-PMP醛亞胺、N-二苯基膦酰基醛亞胺與N-Boc靛紅亞胺均可與3-異硫氰酸酯氧化吲哚1反應得到目標分子.
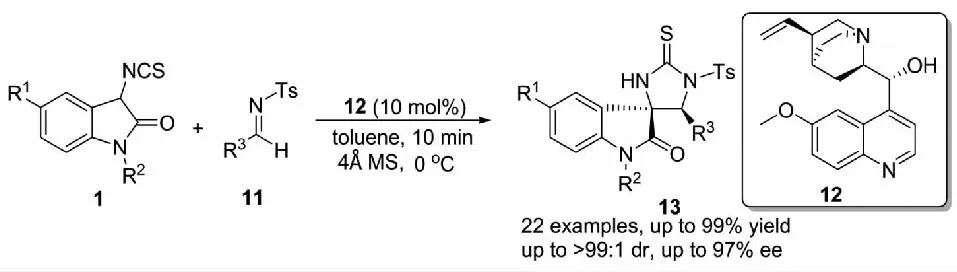
圖7 3-異硫氰酸酯氧化吲哚與醛亞胺的不對稱Mannich/cyclization反應
根據實驗結果,并結合有關α-異硫氰酸酯類化合物與醛亞胺的文獻報道,課題組對該反應提出了可能的過渡態.奎寧C9-OH與Ts保護的亞胺形成氫鍵,奎寧的三級胺部分奪取3-異硫氰酸酯氧化吲哚3位碳原子上的質子,使其烯醇異構化,烯醇化的3-異硫氰酸酯氧化吲哚3位的Si面進攻亞胺的Si面;隨后,氮負離子進攻3-異硫氰酸酯氧化吲哚中NCS基團的碳原子,進而得到螺環氧化吲哚產物13(見圖8).
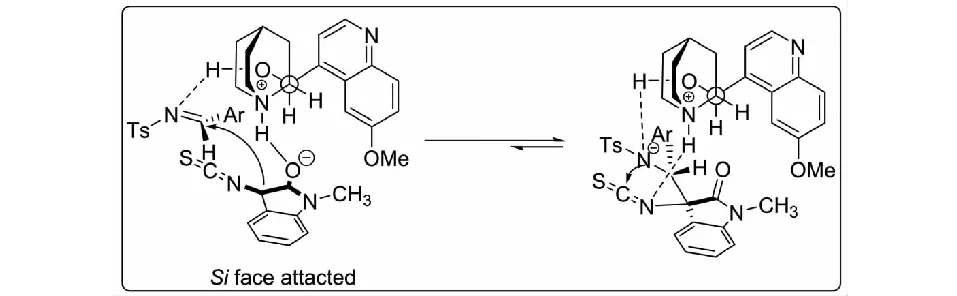
圖8 3-異硫氰酸酯氧化吲哚與醛亞胺可能的反應過渡態
2019年,課題組報道了3-異硫氰酸酯氧化吲哚1與1,3,5-三嗪14的不對稱Mannich/cyclization反應.在奎寧-硫脲15的催化下,1,3,5-三取代的六氫三嗪14原位生成甲醛亞胺14’,與3-異硫氰酸酯氧化吲哚1發生不對稱的Mannich/cyclization反應,以高達99%的收率和93%的ee值得到咪唑烷-2-硫酮螺氧化吲哚的衍生物16(見圖9)[30].這是首次報道有機小分子催化三嗪參與不對稱反應的例子.
Cd在貴州9個地區農業土壤中的吸附呈先快速吸附,然后慢速吸附再到平衡吸附的趨勢,其動力學過程可用偽二級動力學模型較好地描述。其吸附數據可用Freundlich模型較好地擬合。在低濃度區,呈線性增加趨勢,隨著液相中Cd濃度的增加,土壤對Cd的吸附量增大,Cd在土壤中的吸附-解吸過程存在滯后效應。
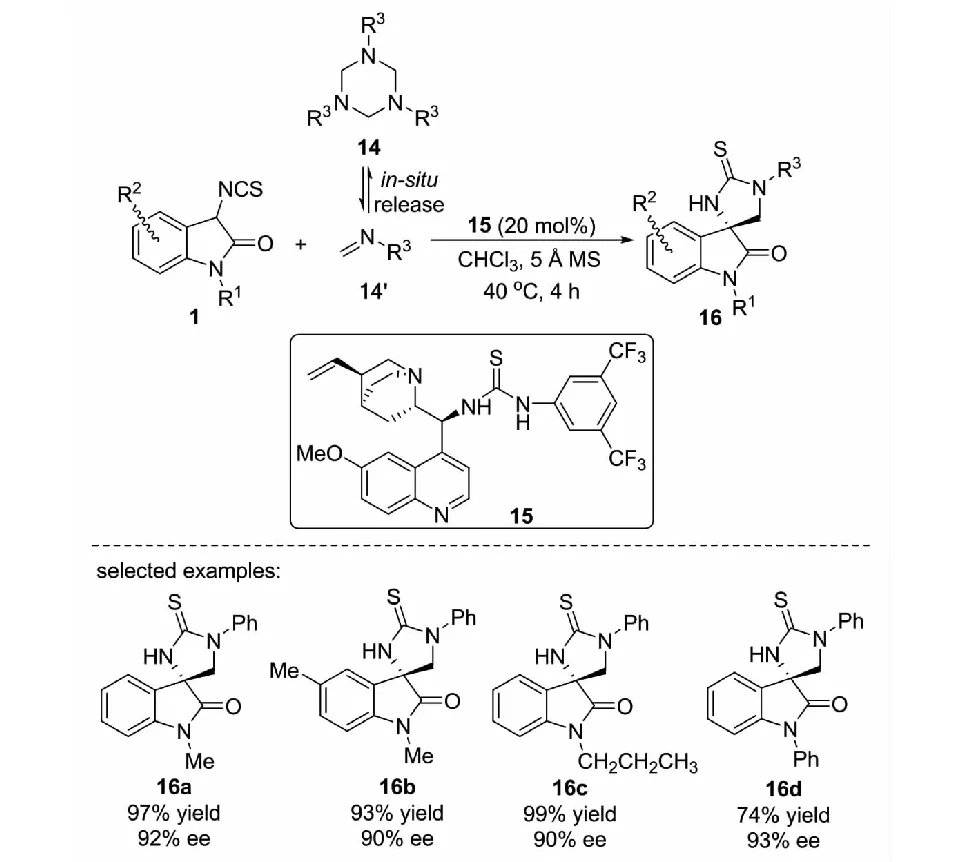
圖9 3-異硫氰酸酯氧化吲哚與三嗪的不對稱Mannich/cyclization反應
此外,課題組提出了一個合理的反應路徑.1,3,5-三取代的三嗪14原位生成甲醛亞胺,環己二胺-硫脲催化劑的雙N-H鍵與醛亞胺形成氫鍵,進而活化底物;同時,催化劑的三級胺部分奪取3-異硫氰酸酯氧化吲哚碳3位的質子,使其親核性增強;在手性催化劑的立體控制下,3-異硫氰酸酯氧化吲哚活化的碳3位進攻醛亞胺的Re面;緊接著,氮負離子進攻3-異硫氰酸酯氧化吲哚的NCS基團發生分子內的環化反應,得到R構型的咪唑烷-2-硫酮螺氧化吲哚化合物(見圖10).
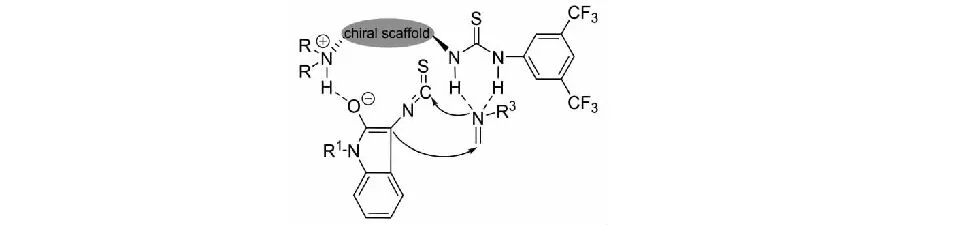
圖10 3-異硫氰酸酯氧化吲哚與1,3,5-三嗪反應可能的活化模式
2020年,課題組又報道了有機小分子催化的3-異硫氰酸酯氧化吲哚1與環酮亞胺17和磺酰亞胺20的不對稱Mannich/cyclization反應,能以高達99%的收率,大于20∶1的dr值和89%的ee值得到一系列手性螺環氧化吲哚衍生物19和22(見圖11)[31].該報道是3-異硫氰酸酯氧化吲哚與環酮亞胺反應構建含有復雜多環體系的螺氧化吲哚類化合物的第一個例子.
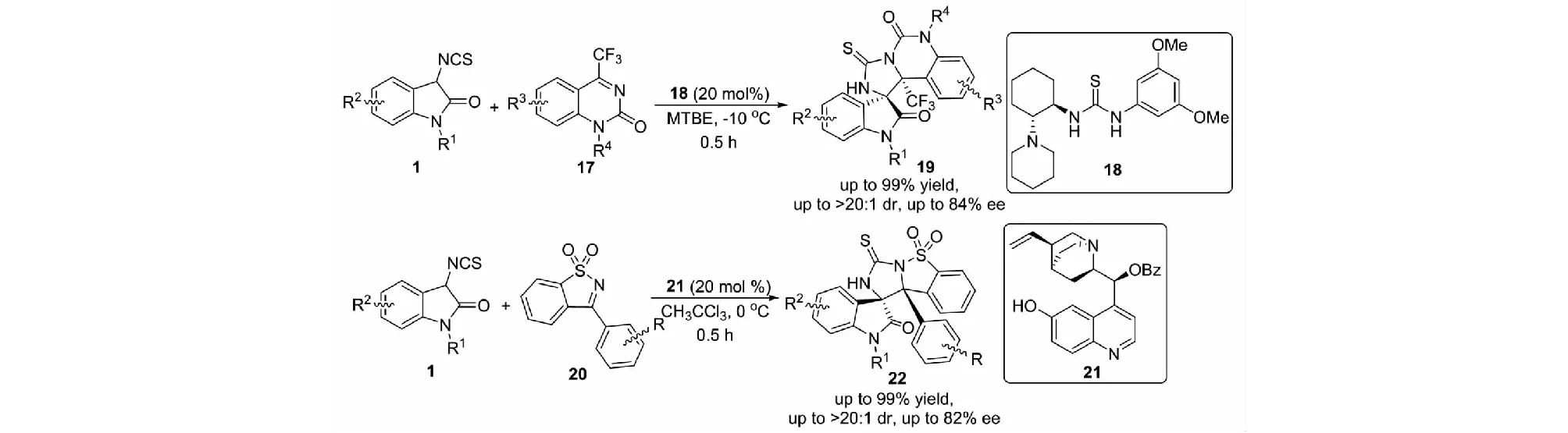
圖11 3-異硫氰酸酯氧化吲哚與環酮亞胺的不對稱Mannich/cyclization反應
根據實驗結果和3-異硫氰酸酯氧化吲哚與亞胺反應的相關報道,課題組提出了可能的反應過渡態.催化劑硫脲部分的雙N-H鍵通過與環酮亞胺形成雙氫鍵來活化環酮亞胺底物;同時,催化劑的叔胺部分奪取3-異硫氰酸酯氧化吲哚碳3位的質子,進而增加其親核性.在環己二胺骨架提供的手性環境下,失去質子的3-異硫氰酸酯氧化吲哚烯醇互變異構體3位的Si面進攻親核試劑環酮亞胺的Si面;隨后,氮負離子進攻3-異硫氰酸酯氧化吲哚NCS基團的碳原子,得到螺環氧化吲哚的化合物(見圖12).
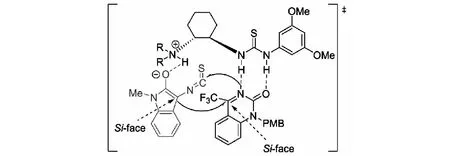
圖12 3-異硫氰酸酯氧化吲哚與環酮亞胺可能的反應過渡態
3 Michael/cyclization反應
3.1 有機小分子催化的Michael/cyclization反應
2013年,課題組報道了奎寧衍生的硫代氨基甲酸酯24催化3-異硫氰酸酯氧化吲哚1與烯基吖內酯23及3-烯基氧化吲哚26的不對稱Michael/cyclization反應,能夠以高達99%的收率,大于99∶1的dr值和大于99%的ee值得到3個連續手性中心的吡咯烷-2-硫酮螺氧化吲哚衍生物25和27(見圖13)[32].
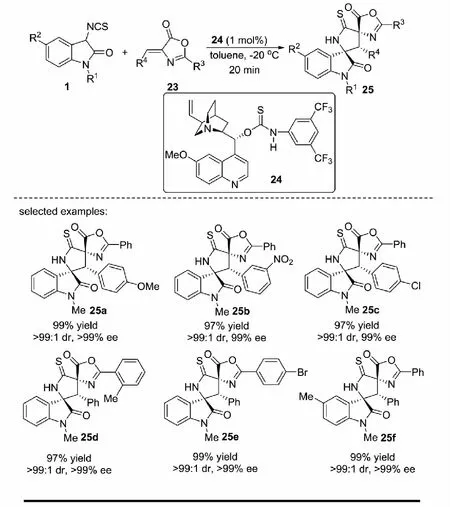
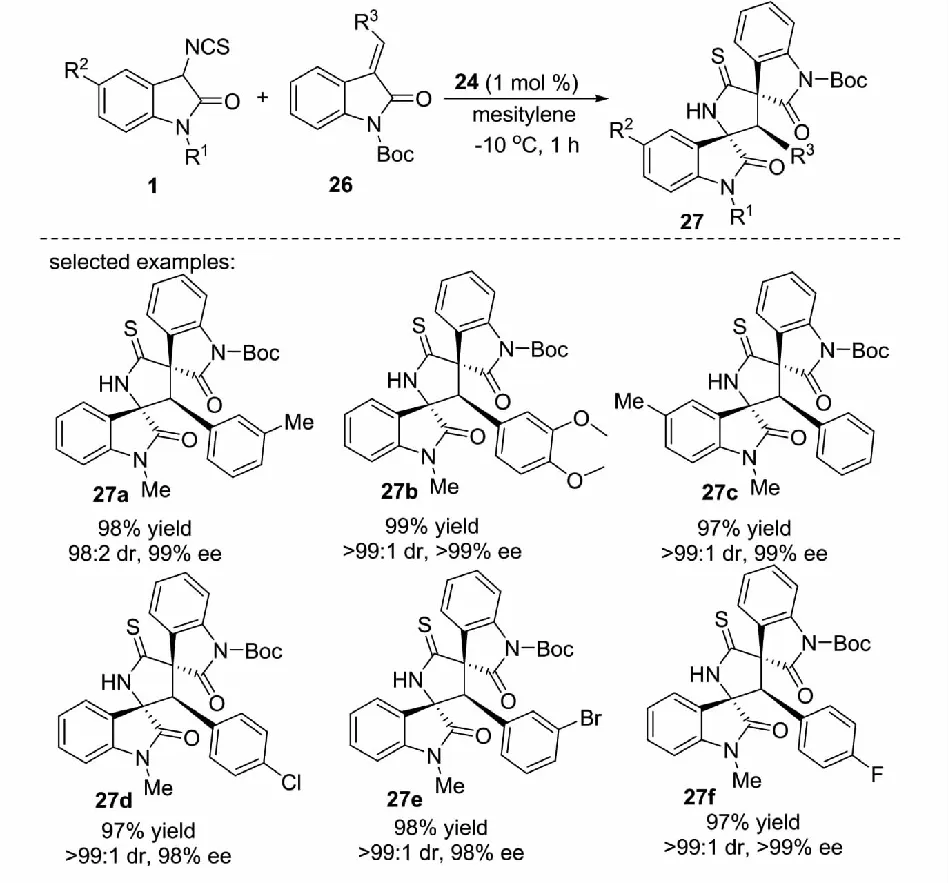
圖13 3-異硫氰酸酯氧化吲哚參與的不對稱Michael/cyclization反應
為了進一步說明3-異硫氰酸酯氧化吲哚在合成結構復雜的螺環氧化吲哚化合物方面的通用性,2013年,課題組報道了商業化的奎寧12催化3-異硫氰酸酯氧化吲哚1與3-甲基-4-硝基-5-烯基異惡唑28之間的Michael/cyclization反應.在溫和的反應條件下,能以高達97%的收率,大于99∶1的dr值和98%的ee值得到吡咯烷-2-硫酮螺氧化吲哚化合物29(見圖14)[33].通過克級反應與產物的衍生化實驗證實了該方法的潛在應用價值.
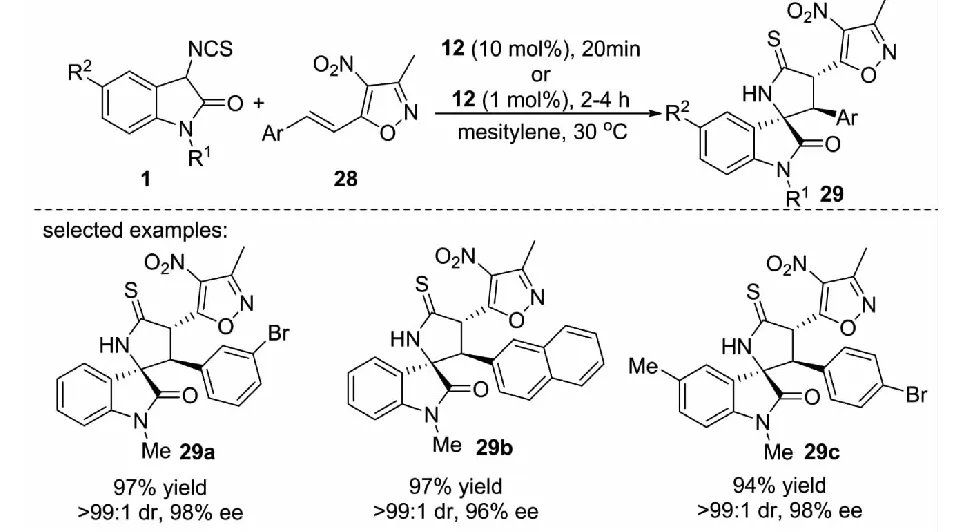
圖14 3-異硫氰酸酯氧化吲哚與3-甲基-4-硝基-5-烯基異惡唑的不對稱反應
基于課題組利用有機小分子催化劑催化3-異硫氰酸酯氧化吲哚參與的Michael/cyclization反應構建吡咯烷-2-硫酮螺氧化吲哚化合物取得的顯著成就,2014年,課題組又報道了3-異硫氰酸酯氧化吲哚1與不飽和吡唑啉酮30及不飽和異惡唑啉酮32的Michael/cyclization反應,仍然以商業化的奎寧12為催化劑,能夠以高收率和良好的立體選擇性得到吡咯烷-2-硫酮螺氧化吲哚衍生物31和33(見圖15)[34].
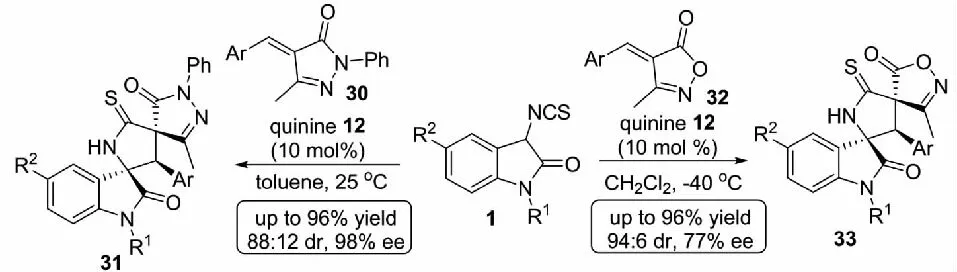
圖15 3-異硫氰酸酯氧化吲哚參與的不對稱Michael/cyclization反應
此外,課題組提出了可能的活化模式.奎寧的三級胺部分奪取3-異硫氰酸酯氧化吲哚3位上的質子,同時,奎寧C9-OH部分通過氫鍵作用活化不飽和吡唑啉酮與不飽和異惡唑啉酮.第一步,3-異硫氰酸酯氧化吲哚失去質子后互變的異構體3位碳負離子進攻不飽和吡唑啉酮與不飽和異惡唑啉酮的β位;緊接著,不飽和吡唑啉酮與不飽和異惡唑啉酮的α位進攻3-異硫氰酸酯氧化吲哚NCS基團的碳原子,進而得到螺環氧化吲哚產物(見圖16).

圖16 3-異硫氰酸酯氧化吲哚與不飽和吡唑啉酮及不飽和異惡唑啉酮反應可能的催化模式
2019年,課題組報道了手性雙功能硫脲催化劑35催化3-異硫氰酸酯氧化吲哚1與β,γ-不飽和α-酮酯34的不對稱Michael/cyclization反應,能夠以高達99%的收率,大于99∶1的dr值和大于99%的ee值得到一系列吡咯烷-2-硫酮螺氧化吲哚衍生物36(見圖17)[35].值得注意的是,在生物活性的初步評估中發現產物螺環氧化吲哚36a和36b具有顯著的抗炎活性.
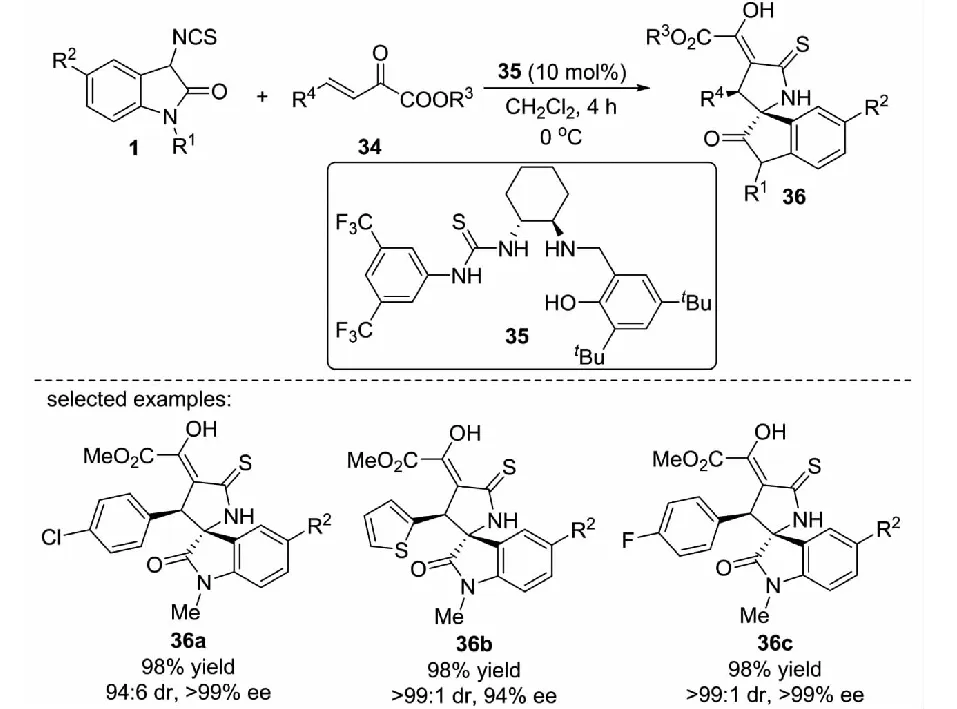
圖17 3-異硫氰酸酯氧化吲哚與β,γ-不飽和α-酮酯的不對稱Michael/cyclization反應
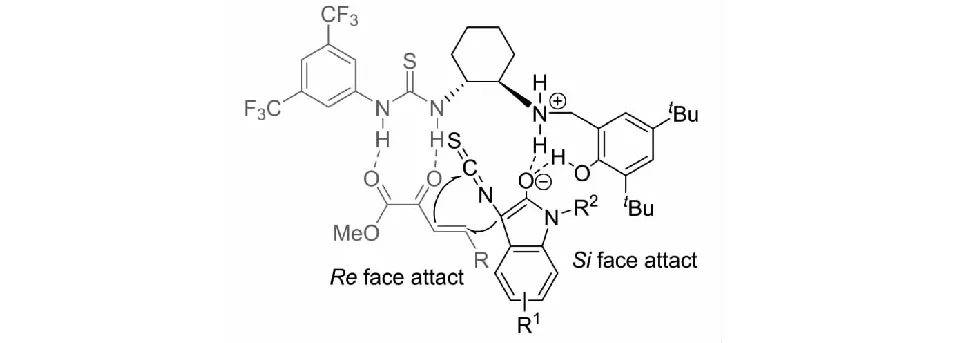
圖18 3-異硫氰酸酯氧化吲哚與β,γ-不飽和α-酮酯可能的反應催化模式
2019年,課題組報道了以商業化的奎寧12催化3-異硫氰酸酯氧化吲哚1與3-甲基-4-硝基-5-吲哚烯基異惡唑37的不對稱Michael/cyclization串聯反應,能夠以高達95%的收率,大于20∶1的dr值和96%的ee值得到3個手性中心相鄰的雙螺環氧化吲哚化合物38.該報道不同于之前報道的3-甲基-4-硝基-5-吲哚烯基異惡唑的α-區域選擇性的不對稱[3+2]環加成反應,它是首例β-區域選擇性的不對稱[3+2]環加成反應(見圖19)[36].
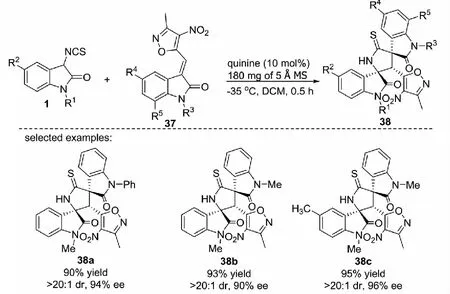
圖19 3-異硫氰酸酯氧化吲哚與3-甲基-4-硝基-5-吲哚烯基異惡唑的Michael/cyclization反應
課題組還提出了可能的活化模式.奎寧的叔胺部分作為堿奪取3-異硫氰酸酯氧化吲哚3位的質子,烯醇互變的異構體2位羥基與叔胺形成氫鍵,奎寧C9-OH與3-甲基-4-硝基-5-吲哚烯基異惡唑的硝基形成氫鍵,進而活化底物.3-異硫氰酸酯氧化吲哚失去質子后互變的異構體3位的碳負離子進攻親電試劑3-甲基-4-硝基-5-吲哚烯基異惡唑的β位;隨后,親電試劑的α位進攻3-異硫氰酸酯氧化吲哚NCS基團的碳原子,最終得到雙螺環氧化吲哚產物(見圖20).
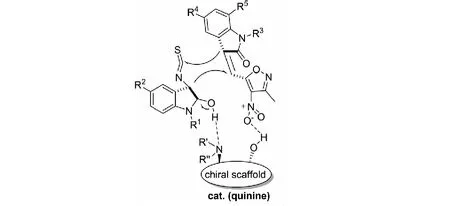
圖20 3-異硫氰酸酯氧化吲哚與3-甲基-4-硝基-5-吲哚烯基異惡唑反應可能的活化模式
3.2 無催化劑的Michael/cyclization反應
2018年,課題組報道了在沒有催化劑條件下,3-異硫氰酸酯氧化吲哚1與香豆素類化合物39和41的Michael/cyclization反應,能夠以高達92%的收率和大于20∶1的dr值得到3個手性中心相鄰且含有3個生物活性基團的化合物40和42(見圖21)[37].值得注意的是,采用羧基活化-脫羧的策略促進化學惰性的香豆素類化合物39和41參與Michael/cyclization反應是非常有效的.此外,該方法可用于擴展生物活性分子的種類及數量,為藥物篩選提供新的候選分子.
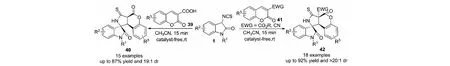
圖21 3-異硫氰酸酯氧化吲哚與香豆素類化合物的Michael/cyclization反應
4 不對稱去芳構化[3+2]環化反應
2015年,課題組報道了奎寧衍生的硫代氨基甲酸酯24催化的3-異硫氰酸酯氧化吲哚1與3-硝基吲哚43的不對稱去芳構化[3+2]環化反應,以高收率和優秀的立體選擇性得到3個手性中心相鄰的手性吲哚啉并吡咯烷2-硫酮螺環氧化吲哚的衍生物44(見圖22)[38].但是,反應體系對于R2為Et、Bn和Ph的3-異硫氰酸酯氧化吲哚及R2為Ms、Ns、Cbz、Ac和Boc的3-硝基吲哚等底物的適用性不佳,反應的立體選擇性差.另外,為了進一步擴大這一方法的應用潛力,對產物進行了衍生化實驗,產物可轉化為結構多樣的螺環氧化吲哚產物(見圖23).更重要的是,這是有機小分子催化3-硝基吲哚參與的不對稱去芳構化環化反應的首例報道.
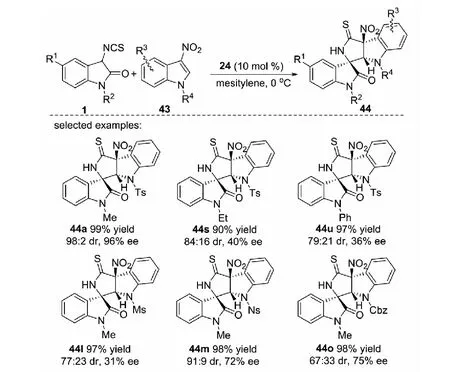
a) MeI,K2CO3,acetone,0 oC,12 h;b) BnBr,K2CO3,acetone,0 ℃,12 h;c) H2O2,HCOOH,CH2Cl2,0 ℃ to room temperature,12 h;d) Zn,TMSCl,EtOH,reflux,4 h;e) AcOH,H2SO4,100 ℃,3 h;f) NaBH4,NiCl2,MeOH,24 h圖22 3-異硫氰酸酯氧化吲哚與3-硝基吲哚的不對稱去芳構化[3+2]環化反應
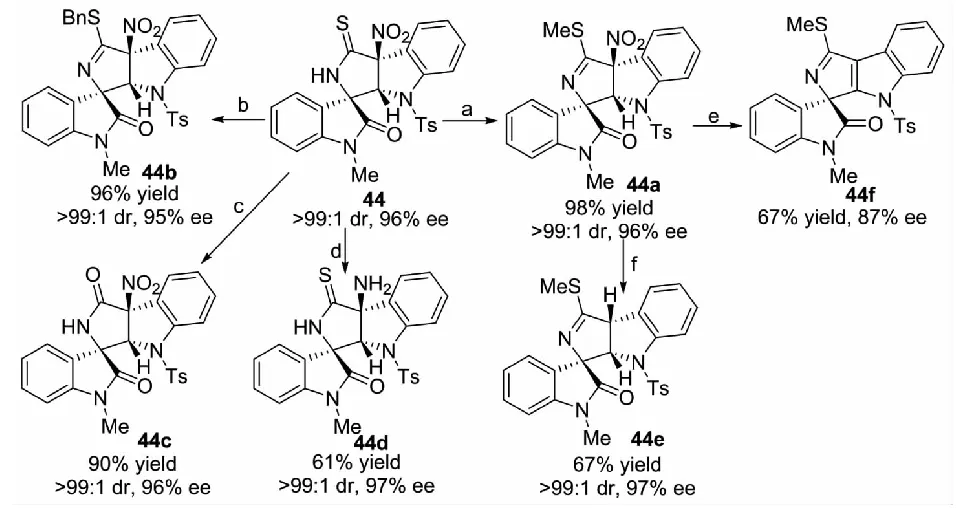
圖23 多環螺環氧化吲哚的衍生實驗
鑒于有機小分子催化的3-異硫氰酸酯氧化吲哚與3-硝基吲哚的不對稱去芳構化[3+2]環化反應在R2為Et、Bn和Ph的3-異硫氰酸酯氧化吲哚及R2為Ms、Ns、Cbz、Ac和Boc的3-硝基吲哚等底物中的適用性不佳,同年,課題組又發展了金屬催化3-異硫氰酸酯氧化吲哚1與3-硝基吲哚43的不對稱[3+2]環化反應.以Zn(OTf)2與二苯胺連接的雙惡唑啉配體L1的絡合物為催化劑,該反應順利進行,以幾乎定量的收率和優秀的立體選擇性得到一系列螺環氧化吲哚類衍生物44(見圖24和表1)[39].值得關注的是,該課題的金屬催化策略在非對映選擇性和對映選擇性方面明顯優于先前報道的有機小分子催化的方法.該催化體系彌補了有機小分子催化體系在立體選擇性方面的不足之處.
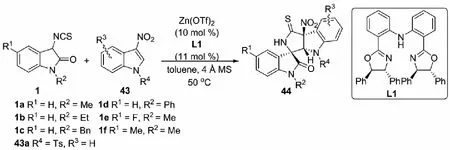
圖24 金屬催化與小分子催化3-異硫氰酸酯氧化吲哚與3-硝基吲哚的不對稱去芳構化[3+2]環化反應
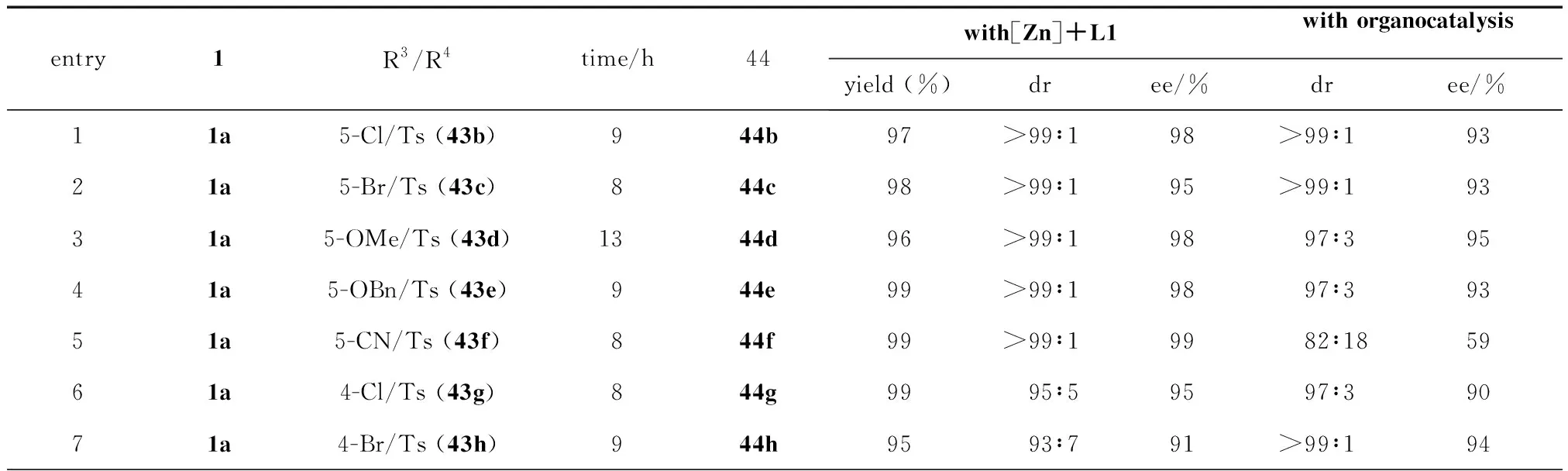
表1 金屬催化與小分子催化3-異硫氰酸酯氧化吲哚與3-硝基吲哚的不對稱去芳構化[3+2]環化反應的對比表
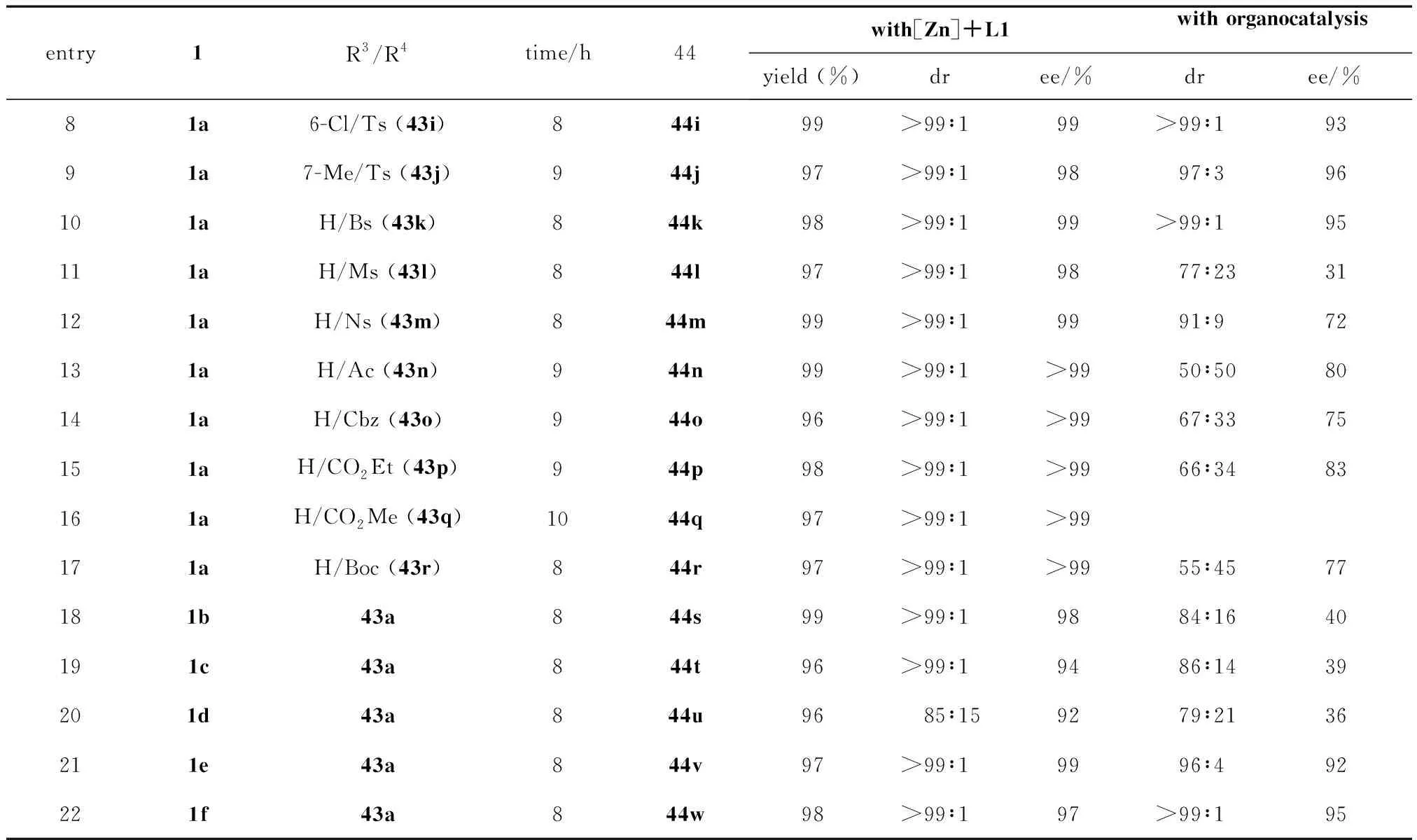
續表1
此外,課題組還提出了一個可能的活化模式.Zn(Ⅱ)作為路易斯酸活化3-硝基吲哚,同時,配體的N-H基團作為路易斯堿活化3-異硫氰酸酯氧化吲哚.首先,3-異硫氰酸酯氧化吲哚3位的Si面進攻3-硝基吲哚2位的Re面;隨后,3-硝基吲哚3位的碳負離子進攻NCS基團的碳原子而發生分子內的環化反應,進而得到3個手性中心相鄰的多環螺氧化吲哚的產物44(見圖25).
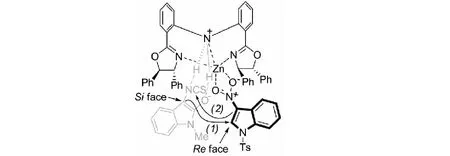
圖25 金屬Zn催化3-異硫氰酸酯氧化吲哚與3-硝基吲哚[3+2]環化反應可能的催化模式
2017年,課題組報道了以Zn(OTf)2與雙惡唑啉配體L2組成的絡合物為催化劑催化3-異硫氰酸酯氧化吲哚1與3-硝基苯并噻吩45及3-硝基吡啶并噻吩46的不對稱去芳構化[3+2]環化反應,以幾乎定量的收率和優秀的對映選擇性得到一系列結構多樣的且3個手性中心相鄰的螺環氧化吲哚化合物47和48(見圖26)[40].
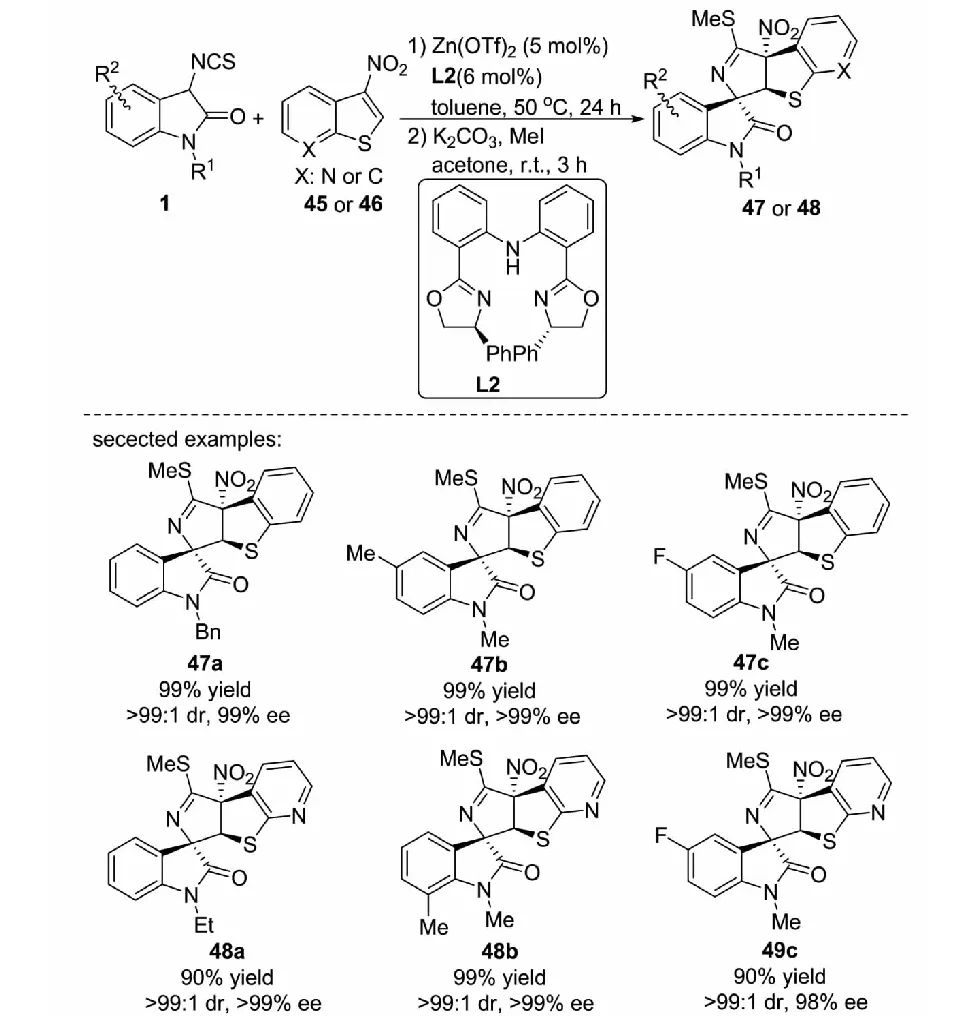
圖26 3-異硫氰酸酯氧化吲哚與3-硝基苯并噻吩及3-硝基吡啶并噻吩的不對稱去芳構化[3+2]環化反應
課題組同樣提出了可能的催化模式,該催化體系為雙活化模式.Zn(OTf)2與雙惡唑啉配體L2組成的絡合物催化劑作為路易斯酸與硝基配位,以活化3-硝基苯并噻吩;催化劑的N-H基團作為路易斯堿活化3-異硫氰酸酯氧化吲哚.3-異硫氰酸酯氧化吲哚3位的Si面進攻3-硝基苯并噻吩2位的Re面;隨后,3-硝基苯并噻吩的3位的碳負離子進攻NCS基團的碳原子而發生分子內的環化反應,得到一系列目標產物(見圖27).
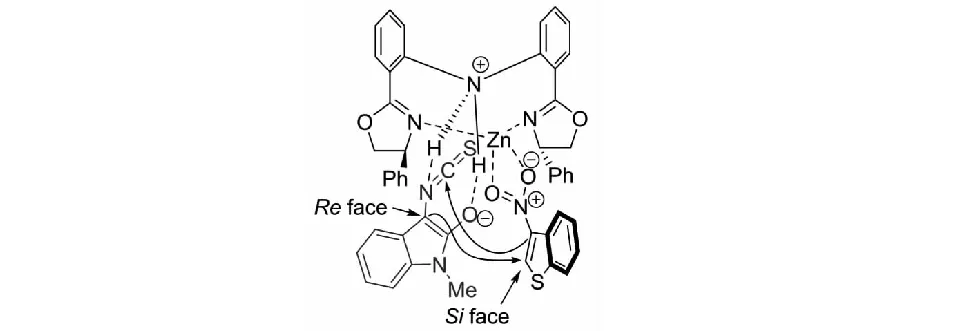
圖27 3-異硫氰酸酯氧化吲哚與3-硝基苯并噻吩反應可能的催化模式
基于這一工作的啟發,課題組對金屬Zn催化的不對稱去芳構化反應進行了更加深入的研究.2018年,課題組報道了金屬Zn(OTf)2與雙惡唑啉配體L2組成的絡合物為催化劑,催化3-異硫氰酸酯氧化吲哚1與2-硝基苯并呋喃49的不對稱去芳構化[3+2]環化反應,以93%~98%的收率 ,大于或等于99∶1的dr值和大于或等于99%的ee值得到結構多樣的含有2,3-二氫苯并呋喃結構單元且有3個相鄰手性中心的螺環氧化吲哚衍生物50(見圖28)[41].
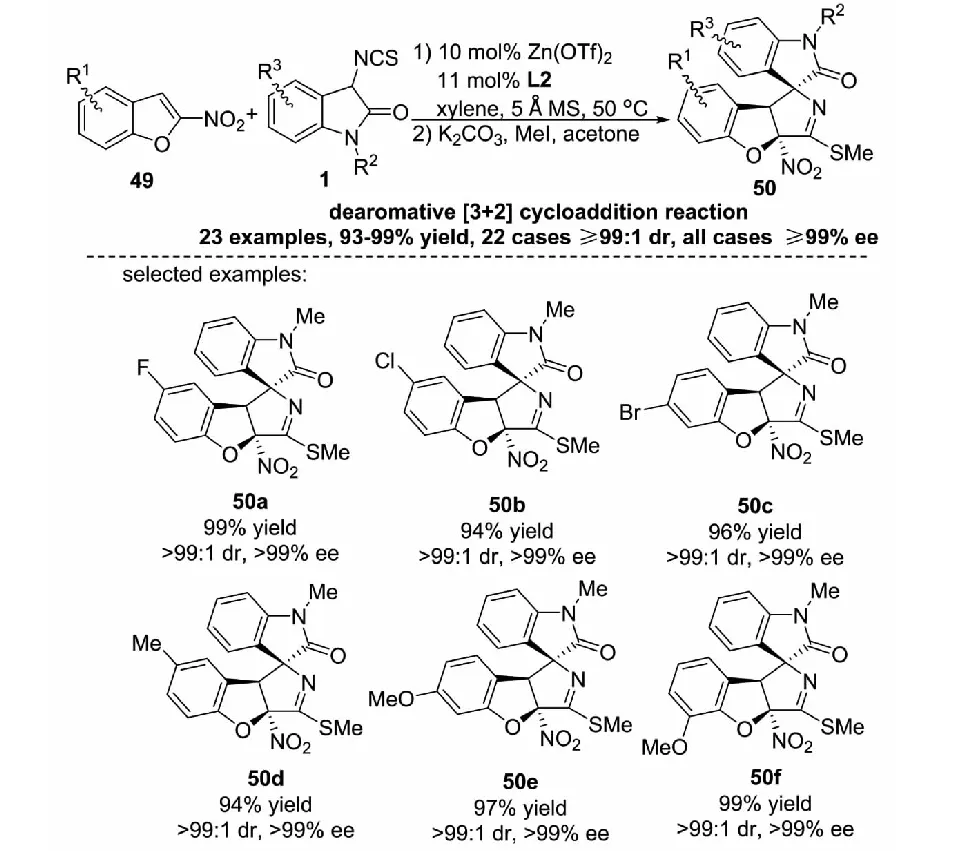
圖28 3-異硫氰酸酯氧化吲哚與2-硝基苯并呋喃的不對稱去芳構化[3+2]環化反應
課題組同樣提出了可能的催化模式.Zn(OTf)2/雙惡唑啉L2組成的絡合物為催化劑,Zn(II) 與2-硝基苯并呋喃49的硝基配位以活化底物,同時,雙(惡唑啉)配體的N-H基團作為路易斯堿活化3-異硫氰酸酯氧化吲哚.在Zn(OTf)2/雙惡唑啉配合物催化劑的雙活化模式下,3-異硫氰酸酯氧化吲哚3位的Re面進攻2-硝基苯并呋喃3位的Re面;隨后,2-硝基苯并呋喃2位的碳負離子進攻NCS基團的碳原子而發生分子內的環化反應,得到高收率和高立體選擇性的目標產物(見圖29).
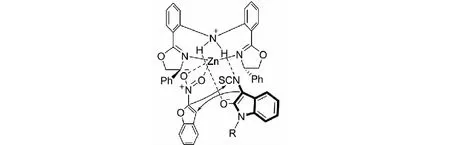
圖29 3-異硫氰酸酯氧化吲哚與2-硝基苯并呋喃反應可能的催化模式
同年,課題組又報道了以金屬Zn(OTf)2與雙惡唑啉配體L2的絡合物為催化劑,3-異硫氰酸酯氧化吲哚1與2-硝基吲哚51及2-硝基苯并噻吩53的不對稱去芳構化環化反應,以幾乎定量的收率和優秀的立體選擇性得到結構多樣且3個手性中心相鄰的螺環氧化吲哚化合物52和54(見圖30)[42]
同樣,課題組提出了可能的催化模式.首先,Zn(OTf)2/雙惡唑啉絡合物的Zn(II)活化2-硝基吲哚或2-硝基苯并噻吩;同時,二苯胺連接的雙(惡唑啉)配體的N-H基團作為路易斯堿活化3-異硫氰酸酯氧化吲哚.在Zn(OTf)2/雙惡唑啉絡合物催化劑的雙活化模式下,3-異硫氰酸酯氧化吲哚3位的Re面進攻2-硝基吲哚或2-硝基苯并噻吩3位的Re面;隨后,2-硝基吲哚或2-硝基苯并噻吩2位的碳負離子進攻NCS基團的碳原子而發生分子內的環化反應,形成一系列螺環氧化吲哚化合物(見圖31).
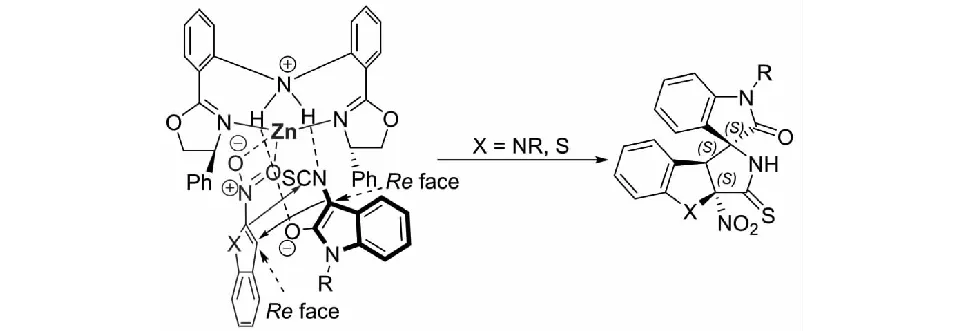
圖31 金屬催化3-異硫氰酸酯氧化吲哚與2-硝基吲哚及2-硝基苯并噻吩反應可能的活化模式
2021年,課題組報道了有機小分子催化的3-異硫氰酸酯氧化吲哚1與缺電子苯并雜環49和53的不對稱去芳構化[3+2]環化反應.以辛可尼丁-(L)苯丙氨酸-硫脲雙功能三氫鍵催化劑55催化反應,能以高達99%的收率,94∶6的dr值和97%的ee值得到結構多樣的含有3個手性中心相鄰的螺環氧化吲哚衍生物50′和54′(見圖32與圖33)[43].以辛可尼丁-芳酰胺56催化3-異硫氰酸酯氧化吲哚與2-硝基吲哚51的反應時,以高收率和良好的立體選擇性得到目標產物52′(見圖34)[43].與課題組先前過渡金屬催化的研究相比,有機小分子催化的3-異硫氰酸酯氧化吲哚1與2-硝基苯并呋喃及2-硝基苯并噻吩反應得到了非對映異構體產物50′和54′,與2-硝基吲哚的反應中獲得了對映異構體產物52′.
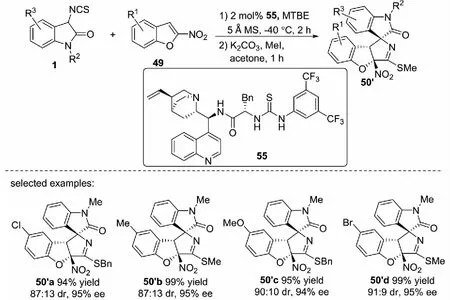
圖32 有機小分子催化的3-異硫氰酸酯氧化吲哚與2-硝基苯并呋喃的不對稱去芳構化[3+2]環化反應
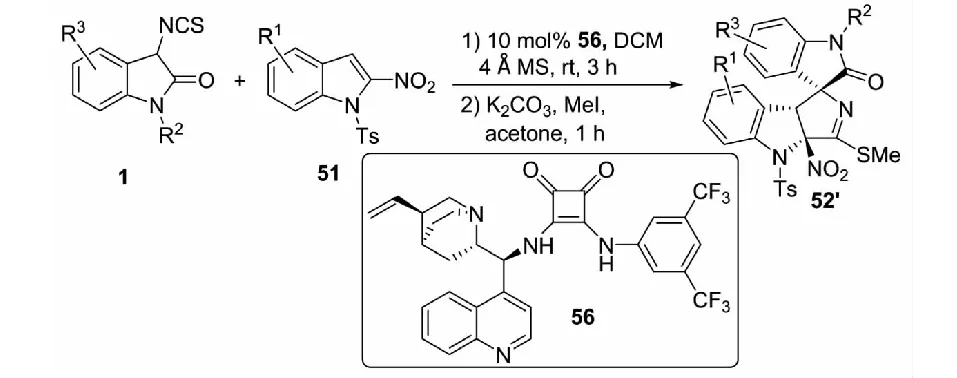
圖34 有機小分子催化的3-異硫氰酸酯氧化吲哚與2-硝基吲哚的不對稱去芳構化[3+2]環化反應
課題組還提出了一種可能的活化模式,解釋了3-異硫氰酸酯氧化吲哚與2-硝基苯并呋喃及2-硝基苯并噻吩之間的不對稱去芳構化環化反應的立體控制.催化劑的3個N-H鍵與2-硝基苯并呋喃或2-硝基苯并噻吩的硝基形成氫鍵,進而被活化和定向;同時,催化劑的叔胺部分活化和定向3-異硫氰酸酯氧化吲哚.在雙活化的催化模式下,3-異硫氰酸酯氧化吲哚C3位的Re面進攻2-硝基苯并呋喃與2-硝基苯并噻吩C3位的Si面,接著進行分子內的環化反應得到(C1R、C2R和C3S)構型的目標產物(見圖35).
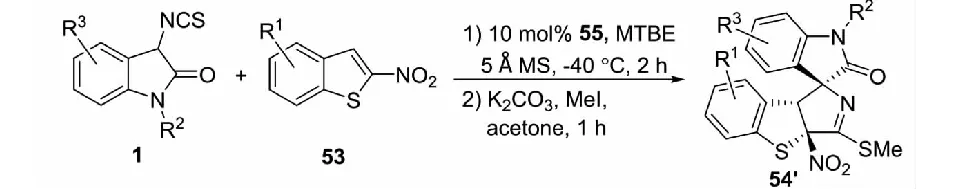
圖33 有機小分子催化的3-異硫氰酸酯氧化吲哚與2-硝基苯并噻吩的不對稱去芳構化[3+2]環化反應
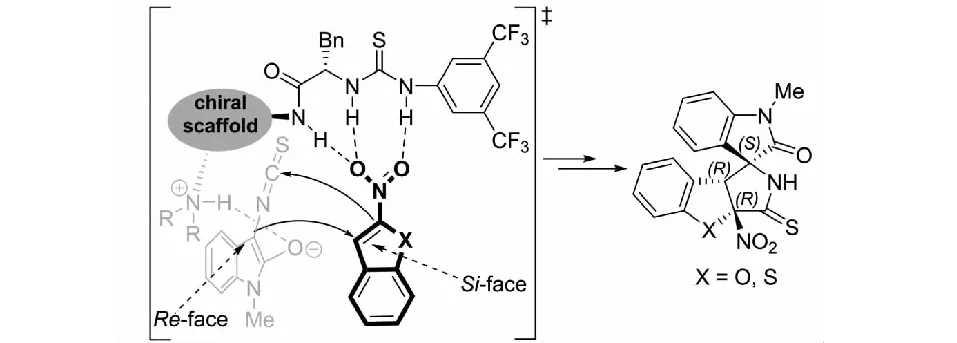
圖35 3-異硫氰酸酯氧化吲哚與2-硝基吲哚可能的反應過渡態
5 [3+3]環化反應
2016年,課題組報道了對甲苯磺酸介導的3-異硫氰酸酯氧化吲哚1與3-吲哚基甲醇57的[3+3]環化反應,能夠以高達99%的收率和大于20∶1的dr值得到一系列螺環氧化吲哚的化合物58(見圖36)[44].此外,提出了可能的催化模式.首先,3-吲哚基甲醇與Br?nsted酸反應,生成水合陽離子化合物A,脫水后得到中間體B;與此同時,3-異硫氰酸酯氧化吲哚3位失去質子后得到烯醇互變的異構體C;接著,C進攻中間體B得到中間體D;最后,吲哚環上2位的碳負離子進攻NCS的碳原子得到目標產物(見圖37).
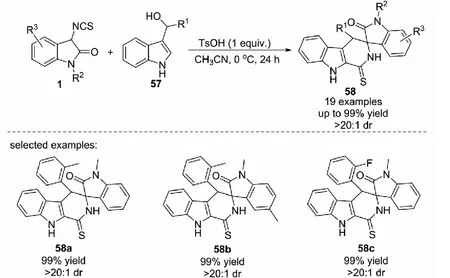
圖36 3-吲哚基甲醇與3-異硫氰酸酯氧化吲哚的[3+3]環化反應
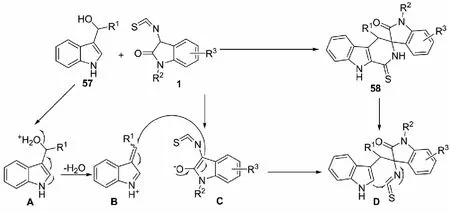
圖37 3-異硫氰酸酯氧化吲哚與3-吲哚基甲醇的 [3+3]環化反應可能的活化模式
6 結 語
在過去的十年中,有機小分子和金屬催化的3-異硫氰酸酯氧化吲哚參與的不對稱串聯環化反應取得了顯著的成果,為制備其他途徑難以合成的螺環氧化吲哚類化合物提供了新方法.然而,目前已發展的3-異硫氰酸酯氧化吲哚參與的串聯環化反應類型仍較局限.未來,開發基于3-異硫氰酸酯氧化吲哚的新型分子間不對稱串聯環化反應將是該領域發展的重要方向,其中包括新型底物的設計合成與新型催化劑體系的設計開發.此外,深入研究反應機理對該領域的進一步發展也至關重要.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
