TET酶家族在神經(jīng)膠質(zhì)瘤干細胞中的分化調(diào)控機制及其潛在治療靶點的研究進展
2022-10-20 02:56:12傅雨昂魏子龍
復(fù)旦學報(醫(yī)學版) 2022年5期
傅雨昂 魏子龍
(1上海市浦東醫(yī)院-復(fù)旦大學附屬浦東醫(yī)院神經(jīng)外科 上海 201399;2復(fù)旦大學公共衛(wèi)生學院2018級預(yù)防醫(yī)學本科生 上海 200032)
TET酶 家 族 蛋 白 由TET1、TET2和TET3組成,都含有一個保守的雙鏈β-螺旋結(jié)構(gòu)域、一個富含半胱氨酸的結(jié)構(gòu)域以及輔助因子Fe(Ⅱ)和α-酮戊二酸的結(jié)合位點,共同形成C端核心催化區(qū)域,TET1和TET3還有一個n端CXXC鋅指結(jié)構(gòu)域,可以與DNA結(jié)合[1]。TET酶家族具有將5-甲基胞嘧啶(5-methylcytosine,5mC)轉(zhuǎn)化為5-羥甲基胞嘧啶(5-hydroxymethylcytosine,5hmC)的能力,進而調(diào)控DNA甲基化[2],不僅調(diào)控細胞分化[3],而且在多種病理生理過程中發(fā)揮重要作用[4]。自TET酶家族蛋白質(zhì)被發(fā)現(xiàn)以來,在神經(jīng)膠質(zhì)瘤以及其他惡性腫瘤中也發(fā)現(xiàn)了5 mC氧化異常和TET酶家族蛋白功能失調(diào)[5-7]。近年來,關(guān)于5mC氧化異常和TET酶家族蛋白功能失調(diào)對神經(jīng)膠質(zhì)瘤的影響得到廣泛的研究[8-10]。
本文對TET酶家族在神經(jīng)膠質(zhì)瘤干細胞(glioma stem cell,GSC)分化過程中,通過去甲基化調(diào)控基因表達的具體機制及其在DNA損傷應(yīng)答和修復(fù)過程的作用加以闡述,并對干預(yù)TET酶家族及其上游異檸檬酸脫氫酶(isocitrate dehydrogenase,IDH)基因的功能治療神經(jīng)膠質(zhì)瘤面臨的挑戰(zhàn)進行分 析 討 論 。 突 變IDH的 拮 抗 劑(mut-IDHantagonist)有望成為新的膠質(zhì)瘤治療策略,介導缺氧條件下DNA修復(fù)相關(guān)的TET1和TET3成為研究熱點,或?qū)⒊蔀樾碌闹委熗黄泣c。
GSC的研究現(xiàn)狀腫瘤干細胞(cancer stem cell,CSC)是具有正常干細胞自我更新、多向分化潛能和無限增殖能力特性的腫瘤細胞[11]。依據(jù)CSC的分子特征,靶向性凋亡CSC或誘導CSC有效分化,是有效控制腫瘤復(fù)發(fā)、轉(zhuǎn)移和耐藥的手段之一。
GSC是神經(jīng)系統(tǒng)腫瘤組織中具有干細胞基本特性的一部分細胞,包括具備自我更新能力、無限增殖能力和多向分化潛能的細胞[12-13],是CSC的熱點研究模型之一。GSC的起源假說之一是正常神經(jīng)干細胞(neural stem cell,NSC)在聚積基因突變、DNA損傷和基因組重排導致的染色體不穩(wěn)定和表觀遺傳修飾混亂后可轉(zhuǎn)化成為CSC[14-15]。GSC起源至今無明確定論,當前研究主要圍繞NSC腫瘤性轉(zhuǎn)化學說、成熟膠質(zhì)細胞失分化學說、融合細胞學說和水平基因傳遞學說等四類學說展開:NSC腫瘤性轉(zhuǎn)化學說認為,GSC可能來源于與之生物學特性相似、基因標志物相同的NSC,或在特定的環(huán)境刺激下通過基因改變獲得自我更新能力的干祖細胞(stem and progenitor cell)[16-19];成熟膠質(zhì)細胞失分化學說則認為其由膠質(zhì)細胞通過失分化、基因突變、基因重排及遺傳改變等方式再次激活,從而具有更強 的 增 殖 能 力 轉(zhuǎn) 變 而 來[16,20];融 合 細 胞 學 說 認 為GSC的來源可能是促融合因子介導的融合細胞[21];水平基因傳遞學說則認為其由膠質(zhì)細胞通過細胞吞噬、吞飲實現(xiàn)與其非子代細胞之間遺傳物質(zhì)的傳遞,使某些致病基因可在不同的細胞之間傳遞而產(chǎn)生[22]。GSC起源于某種細胞還是幾種細胞的合體,NSC轉(zhuǎn)化為GSC的調(diào)控機制和轉(zhuǎn)化發(fā)生的區(qū)域在腦室下區(qū)域還是腦室外區(qū)域尚不明確[23]。
GSC的不對稱分化導致其具有高度異質(zhì)性的特點。早 在2010年,Verhaak等[25]便獲得了腫瘤間異質(zhì)性的證據(jù),根據(jù)不同患者腫瘤樣本中的基因突變和表達譜將膠質(zhì)母細胞瘤(glioblastoma multiforme,GBM)分為4種亞型,其中2種亞型有助于GBM治療后的異常生長和腫瘤復(fù)發(fā)[24-25]。2014年,Sottoriva等[26]通過單細胞測序發(fā)現(xiàn),同一腫瘤組織的不同區(qū)域呈現(xiàn)不同的分型,稱為腫瘤內(nèi)異質(zhì)性,并且不同亞群的GSC對不同的表觀修飾抑制劑具有特異的敏感性[27]。這些研究揭示了不僅腫瘤組織異質(zhì)性會影響治療手段和效果,腫瘤組織GSC所具有的表觀修飾異質(zhì)性也將成為基礎(chǔ)研究和臨床精準治療的關(guān)鍵因素。
TET酶家族的研究現(xiàn)狀TET酶家族蛋白質(zhì)具有保守的富胱氨酸結(jié)構(gòu)域和雙鏈β螺旋結(jié)構(gòu)域,這是依賴Fe(Ⅱ)和α-酮戊二酸的雙加氧酶的催化中心。TET酶家族蛋白質(zhì)具有獨特的酶促功能,可促進DNA去甲基化過程。TET介導5mC氧化,是將其轉(zhuǎn)化為5hmC的雙加氧酶,并進一步氧化產(chǎn)生5-甲酰基胞嘧啶(5-formylcytosine,5fC)和5-羧基胞嘧啶(5-carboxylcytosine,5caC)[28-31],胸腺嘧啶DNA糖苷酶能夠氧化5fC和5caC,再經(jīng)過堿基切除修復(fù)得到未被修飾的胞嘧啶。
TET酶介導的DNA去甲基化受到多種輔助因子和代謝物的調(diào)節(jié)。TET酶的作用依賴于α-KG和Fe2+。α-KG是三羧酸循環(huán)的重要中間體,推測能夠通過降低α-KG的代謝活動抑制TET酶介導的去甲基化作用,當IDH1和IDH2發(fā)生突變時,α-KG無法正常合成,已有的α-KG被轉(zhuǎn)化為2-羥基戊二酸(2-hydroxyglutarate,2HG),2HG是α-KG的類似物,能與α-KG競爭抑制TET蛋白。Fe2+則能增強TET的 催 化 活 性[32]。 鋅 指 轉(zhuǎn) 錄 因 子(zinc finger transcription factors,ZSCAN)可募集TET酶家族,從而調(diào)控包括重編碼在內(nèi)的多種細胞過程,鋅指轉(zhuǎn)錄因子成員中的Zscan4f是TET2調(diào)控靶基因和促進誘導多能干細胞(induced pluripotent stem cell,iPSC)生成的重要伙伴[33]。TET酶家族中TET2易位突變最早在急性髓系白血病中被發(fā)現(xiàn),這一突變導致TET2喪失去甲基化功能。TET酶家族的抑癌機制除了通過去甲基化途徑調(diào)控基因表達,還可能參與了DNA損傷修復(fù)過程。
TET酶家族的3個成員參與DNA損傷應(yīng)答和修復(fù)過程的現(xiàn)象及機制研究分別被報道過。TET1通過ATM依賴的方式對DNA損傷產(chǎn)生響應(yīng),從而在浦肯野細胞內(nèi)產(chǎn)生大量的5hmC修飾,缺失TET1會影響損傷修復(fù)的激活,導致細胞凋亡[34]。TET2有助于5hmC的富集,一種通過與共激活因子和序列特異性DNA結(jié)合因子的三元相互作用將TET2靶向到特定啟動子:Smad核內(nèi)相關(guān)蛋白1(Smad nuclear interacting protein 1,SNIP1)溝 通TET2與包括c-myC在內(nèi)的多種轉(zhuǎn)錄因子的相互作用;TET2以SNIP1依賴的方式保護細胞免受DNA損傷誘導的凋亡;TET2-SNIP1-c-myC軸支持TET2的DNA序列特異性募集。TET2-SNIP1-cmyC通路介導DNA損傷修復(fù)反應(yīng),保護細胞免受DNA損傷誘導的凋亡,從而將表觀遺傳控制與基因組穩(wěn)定性的維持聯(lián)系起來[35]。TET3產(chǎn)生5hmC參與損傷響應(yīng)則被認為是受到ATR磷酸化的調(diào)控,并促進DNA去甲基化和5hmC的積累[36],從而起到DNA修復(fù)與基因組穩(wěn)定性的維護作用[37]。與TET2類似,TET3的催化活性對DNA正常修復(fù)和細胞存活也很重要。有研究曾在TET2和TET3雙敲除的白血病模型中觀察到非同源末端修復(fù)(nonhomologous end joining,NHEJ)和 同 源 重 組 修 復(fù)(homologous recombination,HR)的基因普遍受到轉(zhuǎn)錄抑制。
有研究認為TET酶在細胞復(fù)制過程中有著維持基因組完整性的作用。活性DNA去甲基化可能特異性發(fā)生在DNA損傷部位,這一事實表明TET酶和/或5hmC可能在DNA修復(fù)中起 直 接 作用[38]。5hmC參與的表觀遺傳調(diào)控的缺失可能有助于疾病進展[39-40]。5hmC可能具有DNA損傷的表觀遺傳標記的作用,并被證明可以促進基因組的穩(wěn)定性。
5hmC在DNA修復(fù)中的潛在作用亟待研究,5hmC標記可出現(xiàn)在DNA損傷部位的DNA共價修飾,研究TET活化與5hmC的產(chǎn)生是否與DNA損傷類型有關(guān)至關(guān)重要。5hmC可能是TET在DNA損傷部位激活的副產(chǎn)物,這是清除5mC所必需的。還有一種可能,即對主導DNA修復(fù)的是TET酶,而不是5hmC。確認DNA損傷介導的5hmc識別因子與TET酶家族相互作用的關(guān)系,對于理解5hmC和TET蛋白在DNA修復(fù)中的潛在作用至關(guān)重要[36]。
膠質(zhì)瘤及GSC中涉及到的TET酶家族的研究情況TET酶家族的抑癌機制除了通過去甲基化途徑調(diào)控基因表達,可能還參與了DNA損傷修復(fù)過程。在TET酶家族中存在潛在的勞動分工。
已有研究報道,神經(jīng)膠質(zhì)瘤中的整體低甲基化和位點特異性異常甲基化以及其他表觀遺傳修飾是神經(jīng)膠質(zhì)瘤進展過程中基因組不穩(wěn)定的重要因素。在以缺氧介導的神經(jīng)膠質(zhì)瘤細胞DNA去甲基化過程中,發(fā)揮作用的去甲基化酶是TET1和TET3,而不是TET2。TET1和TET3與八聚體結(jié)合轉(zhuǎn)錄因子4(octamer-binding transcription factor 4,OCT4)和轉(zhuǎn)錄因子NANOG有活性的調(diào)控區(qū)域直接結(jié)合去甲基化酶TET1和TET3。從GSC中敲除去甲基化酶TET1和TET3時,干細胞基因OCT4和NANOG的表達均下降,并抑制神經(jīng)球的形成,在缺氧狀態(tài)下TET1和TET3的去甲基化是OCT4和NANOG過表達的機制,有助于膠質(zhì)瘤中GSC的形成[41]。TET1和TET3在低氧條件下各自獨立調(diào)節(jié)多能性相關(guān)基因和分化相關(guān)基因。
TET2在 產(chǎn) 生5fC和5caC方 面 作用 更 強[30],尤其是增強子上的5hmC富集所必需的[42]。TET2的表達還與GSC對DNA損傷后的修復(fù)相關(guān)。GSC比NSC表達更高水平的DNA修復(fù)基因,這可能是其對輻射或化療誘導的DNA損傷抗性的原因之一。使用siRNA敲除TET2可使GSC細胞系對博萊霉素治療敏感,并且TET2表達的GSC越高則越能抵抗博萊霉素造成的DNA損傷并繼續(xù)增殖,表明TET2確實能促進DNA損傷后的修復(fù)。TET2也可能在細胞周期中催化生成和DNA損傷應(yīng)答相關(guān)的基因啟動子上的5fC/5caC,這些基因在GSC中表達高于NSC,且與TET2水平呈正相關(guān),提示TET2可能調(diào)控這些位點的表達。
TET3蛋白同時分布于細胞核和細胞質(zhì)內(nèi),與GSC中 分 化 表 型(glial fibrillary acidic protein,GFAP)基因的表達高度相關(guān)。人們通過研究GSC核受體TLX(NR2E1)和TET3的級聯(lián)調(diào)控機制發(fā)現(xiàn)TET3可以抑制GSC的生長[43]。TLX是一種可以抑制TET3表達的轉(zhuǎn)錄因子,TET3在TLXTET3軸中處于TLX的下游,可以抑制GSC的生長、自我更新和腫瘤發(fā)生。然而,TET3一方面受到TLX調(diào)控,抑制GSC的生長,另一方面卻可以通過其他通路增加GSC的致瘤性。GCS的細胞外基質(zhì)(extracellular matrix,ECM)通過層黏素-整合素 α6上調(diào)TET3蛋白。TET3反過來介導DNA胞嘧啶5'-羥甲基化(5hmC)的表達,并上調(diào)對維持GSC必要生命活動至關(guān)重要的基因。激活整合素α6-局部黏著斑激酶通路(integrin α6-focal adhesion kinase pathway)可提高GSC中信號傳導與轉(zhuǎn)錄激活因子家 族(signal transduction and activators of transcription,STAT)的 成 員STAT3活 性 ,增 強TET3表達,從而提高5hmC水平。此外,STAT3直接調(diào)控TET3的表達,這兩個蛋白質(zhì)在GSC集群中 與5hmC共 定 位 。 因 此 ,ECM-整 合 素 α6-STAT3-TET3軸調(diào)控了GSC重要基因的羥甲基化,從而增加了GSC的致瘤性[44]。
此外,GSC中TET高表達可能會增加化療耐藥性。GSC在腫瘤中以小亞群形式存在,但積極地促進了整體放療和化療耐藥以及腫瘤復(fù)發(fā)。在多個人源性組織異種移植(patient-derived xenograft,PDX)GSC中分析了全基因組DNA表觀遺傳修飾、增強子標記和轉(zhuǎn)錄組,發(fā)現(xiàn)在GSC中TET表達缺失與5mC和5hmC的整體損失和5fC/5caC的增加相關(guān),與NSC相比,TET表達在GSC分化過程中有不同的反應(yīng),TET3亞細胞定位與分化能力有關(guān);分化誘導的5mC和5hmC重編碼定位于不同的增強子區(qū)域,有助于發(fā)育基因的調(diào)控。整合素α6-STAT3-TET3軸本身也可以增加GSC的耐藥性。GSC中TET2高水平表達有助于化療耐藥,可能是通過調(diào)節(jié)DNA損傷反應(yīng)和修復(fù)系統(tǒng)來實現(xiàn)。
干預(yù)TET酶家族治療神經(jīng)膠質(zhì)瘤的展望TET1活性缺失與多種腫瘤的進展、轉(zhuǎn)移和患者生存率低直接相關(guān),因此TET酶家族通常被認為是抑制腫瘤發(fā)生發(fā)展的抑癌基因的重要表達產(chǎn)物之一。
IDH突變干擾TET酶家族的功能,其所造成的膠質(zhì)瘤CpG島甲基化表型(cytosine-phosphateguanine island methylator phenotype,G-CIMP)與GBM亞型有關(guān),包括在全基因組范圍內(nèi)調(diào)節(jié)甲基化模式,改變轉(zhuǎn)錄程序和改變分化狀態(tài)[45]。當TET酶功能發(fā)生紊亂時,產(chǎn)生的5hmC水平降低,同時縮短患者生存期[46]。因此,TET介導的DNA去甲基化過程及其表觀修飾產(chǎn)物是治療和精準診斷GBM的關(guān)鍵切入點(圖1)。
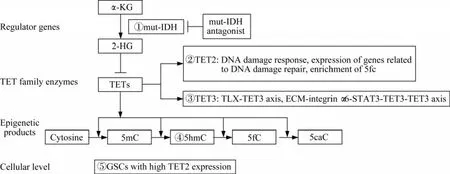
圖1 干預(yù)TET酶家族治療神經(jīng)膠質(zhì)瘤的治療靶點示意圖(治療靶點①~⑤)Fig 1 Schematic diagram of therapeutic targets for intervention of TET family enzymes in the treatment of glioma(therapeutic target①-⑤)
TET2在GSC中的表達高于NSC,TET2的表達與修復(fù)基因的表達呈線性相關(guān)。且TET2與DNA損傷響應(yīng)、修復(fù)相關(guān)基因的表達及其啟動子甲酰化胞嘧啶(5fC)的富集顯著相關(guān),從而成為一種潛在的治療靶點。明確GSC中獨特的表觀基因組特征和相關(guān)的基因網(wǎng)絡(luò),針對這些特征可以在不損傷鄰近正常細胞的情況下應(yīng)用于臨床靶向治療[47]。
調(diào)節(jié)TET3相關(guān)通路可以為治療GBM開辟新的思路。調(diào)節(jié)ECM-整合素α6-STAT3-TET3軸可以逆轉(zhuǎn)通常與CSC增加有關(guān)的耐藥性[44]。調(diào)節(jié)TLX-TET3軸,抑制核受體TLX的表達可抑制GBM的 生 長[43]。 病 毒 載 體 遞 送 的TLX shRNA或納米載體遞送的TLX siRNA治療人類GSC移植小鼠,可抑制腫瘤發(fā)展并延長生存期。
GSC在轉(zhuǎn)錄、DNA修飾和增強子標記等多個水平上存在異質(zhì)性,這些異質(zhì)性也部分反映在GSC間TET表達水平上。高TET2表達的GSC可能是臨床上抗藥物治療的靶點。GSC特異性特征可能賦予該細胞群獨特的分子譜和表觀遺傳特性,并作為抑制或消除GSC群體的潛在治療靶點[41]。
有研究表明,在體外用谷氨酸替代IDH1 132殘基上的精氨酸會造成TET酶活性下降[48]。突變未必導致IDH1蛋白失活,例如阻止使用異檸檬酸作為底物的突變蛋白質(zhì),可以允許酶使用其他未知底物,從而使得酶并未完全失活。如果未來的研究證實了這種可能性,突變的IDH基因可能成為治療干預(yù)的目標。
結(jié)語綜上所述,TET酶家族與神經(jīng)膠質(zhì)瘤發(fā)生相關(guān),TET介導的DNA去甲基化過程及其表觀修飾產(chǎn)物是治療和精準診斷GBM的關(guān)鍵切入點。確定神經(jīng)GSC中獨特的表觀基因組特征和相關(guān)的基因網(wǎng)絡(luò)以及TET2對基因網(wǎng)絡(luò)的調(diào)控機制,可以在不損傷鄰近正常細胞的情況下應(yīng)用于臨床靶向治療。雖然已對TET2基因突變進行了一系列深入研究,但仍需要考慮TET2能否像其他基因一樣作為神經(jīng)膠質(zhì)瘤的危險分層因子。調(diào)節(jié)TET3相關(guān)通路可以降低GSC的致瘤性,抑制腫瘤生長。GSC在轉(zhuǎn)錄、DNA修飾和增強子標記等多個水平上的特異性特征可能賦予該細胞群獨特的分子譜和表觀遺傳特性,作為抑制或消除GSC群體的潛在治療靶點。IDH的突變是腦膠質(zhì)瘤CIMP的分子基礎(chǔ),其所造成的基因組高甲基化狀態(tài)有助于改變轉(zhuǎn)錄程序和分化狀態(tài),為理解神經(jīng)膠質(zhì)瘤的發(fā)生提供了框架,可能成為治療干預(yù)的目標[49]。近年來隨著IDH1/2突變的致癌功能及其相關(guān)代謝產(chǎn)物順-2-羥基戊二酸、缺氧反應(yīng)和DNA修復(fù)研究逐步深入,突變IDH的拮抗劑有望成為新的膠質(zhì)瘤治療策略,與介導缺氧反應(yīng)下DNA修復(fù)相關(guān)的TET1和TET3可能成為新的突破點。
作者貢獻聲明傅雨昂 文獻調(diào)研和整理,論文撰寫和修訂。魏子龍 論文指導和修訂。
利益沖突聲明所有作者均聲明不存在利益沖突。
