硅氧烷預聚體改性熱塑性酚醛樹脂的交聯(lián)結(jié)構(gòu)及其力學性能
2022-11-13 07:39:14徐力吳謙秋雷子萱李嘉玄劉育紅
化工學報 2022年10期
徐力,吳謙秋,雷子萱,李嘉玄,劉育紅
(西安交通大學化學工程與技術(shù)學院,陜西 西安 710049)
引 言
酚醛樹脂是世界上最早實現(xiàn)工業(yè)化生產(chǎn)的合成樹脂,其原料廉價易得,生產(chǎn)設(shè)備簡單,憑借優(yōu)異的耐熱性、電絕緣性、阻燃性能、力學性能和化學穩(wěn)定性,在航空、航天、電子、交通運輸?shù)雀鱾€領(lǐng)域得到了廣泛的應(yīng)用[1-4]。但是,酚醛樹脂的分子結(jié)構(gòu)上存在大量與亞甲基相連的芳環(huán)結(jié)構(gòu)以及苯環(huán)上的酚羥基,使得酚醛樹脂脆性大和易氧化,限制了它在高性能領(lǐng)域的應(yīng)用[5-9]。因此,提高酚醛樹脂的韌性和耐熱性是亟待解決的問題。
外增韌可以有效地改善酚醛樹脂的韌性,通過物理混合向酚醛樹脂中引入橡膠彈性體、剛性納米粒子等改性劑,彈性體自身鏈段的柔韌性、微納粒子的尺寸效應(yīng),在樹脂斷裂過程中起到阻止裂紋擴展、吸收應(yīng)力等作用,從而顯著提高樹脂的韌性[10-15]。但是,橡膠類彈性體在增韌酚醛樹脂的同時,會降低樹脂的楊氏模量、耐熱性以及玻璃化轉(zhuǎn)變溫度(Tg)等關(guān)鍵性能[16-18]。納米粒子的引入雖然可以提高韌性,但其與酚醛樹脂界面相容性差、易團聚等缺點限制了其增韌效果[19]。因此,協(xié)同提高酚醛樹脂的韌性和耐熱性仍是一大挑戰(zhàn)。有機硅聚合物由于其主鏈的Si—O—Si 鍵能較高,分子鏈柔性大且熱穩(wěn)定性高,成為了協(xié)同提高酚醛樹脂韌性和耐熱性的備選材料[20-21]。但是有機硅鏈段與酚醛樹脂相容性差,其改性的酚醛樹脂往往很難達到預期的耐熱性和力學性能。為了解決這一問題,現(xiàn)有的研究是在有機硅鏈段中引入增容結(jié)構(gòu)或硅烷偶聯(lián)劑提高其與酚醛樹脂的相容性,從而使得在固化過程中,有機硅聚合物與酚醛樹脂形成接枝或嵌段共聚物,提高增韌的效果[22-23]。
近幾年來,研究人員提出了一種將部分反應(yīng)的亞結(jié)構(gòu)(partially reacted substructure,PRS)引入到反應(yīng)的樹脂混合物中,合成一系列具有相同化學組成但分子鏈段排列不同的聚合物網(wǎng)絡(luò)的方法,從而實現(xiàn)增韌的目的[24-27]。受此啟發(fā),本研究合成一種環(huán)氧化硅氧烷(ES),采用DSC、紅外及流變法探究不同預聚程度的PES 改性熱塑性酚醛樹脂的固化行為,特別是從固化過程中物理狀態(tài)(凝膠態(tài)和玻璃態(tài))調(diào)控的角度出發(fā),構(gòu)建不同拓撲結(jié)構(gòu)的交聯(lián)網(wǎng)絡(luò),在此基礎(chǔ)上,分析樹脂的組成、ES的自聚程度對改性酚醛樹脂交聯(lián)網(wǎng)絡(luò)、熱穩(wěn)定性以及力學性能的影響。
1 實驗材料和方法
1.1 材料
乙酸乙酯(分析純),購自天津恒興化學試劑制造有限公司;4-乙烯環(huán)氧環(huán)己烷(分析純)、1,1,5,5-四甲基二苯基三硅氧烷(分析純)、咪唑(分析純)、1,3-二乙烯基-1,1,3,3-四甲基二硅氧烷鉑(2%二甲苯溶液),購自上海麥克林生化科技有限公司;無水乙醇(分析純),購自天津富宇精細化工有限公司;熱塑性酚醛樹脂(工業(yè)級),購自山東圣泉新材料股份有限公司;六次甲基四胺(分析純),購自上海阿拉丁生化科技股份有限公司;正己烷(分析純),購自廣東光華科技股份有限公司。
1.2 合成路線
1.2.1 環(huán)氧化硅氧烷(ES)的合成 ES 的合成反應(yīng)路徑如圖1所示。
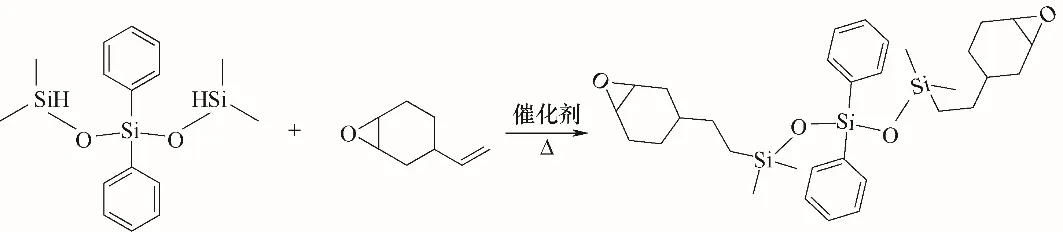
圖1 ES的合成反應(yīng)Fig.1 Synthesis of ES
(1)配制A 液:取20.00 g 的1,1,5,5-四甲基二苯基三硅氧烷加入到40 ml乙酸乙酯中。配制B液:取16.52 g的4-乙烯環(huán)氧環(huán)己烷和0.17 g催化劑1,3-二乙烯基-1,1,3,3-四甲基二硅氧烷鉑的2%二甲苯溶液加入到50 ml乙酸乙酯中。
(2)在氮氣氛圍,60℃條件下,利用恒壓漏斗將A 液緩慢滴加到裝有B 液的三口燒瓶中,滴加完成后,升溫至80℃,恒溫攪拌回流反應(yīng)8 h。
(3)反應(yīng)結(jié)束后,對三口燒瓶中剩余溶液進行旋轉(zhuǎn)蒸發(fā),除去溶劑和部分未反應(yīng)的小分子物質(zhì),得到粗產(chǎn)物。
(4)利用層析柱對所得粗產(chǎn)物進行提純處理,最終得到淺黃色透明液體,即為環(huán)氧化硅氧烷(ES)。
1.2.2 ES 與NR 共混預聚體的制備 稱取20.00 g
熱塑性酚醛樹脂(NR)于燒杯中,加入20.00 g 無水乙醇使樹脂充分溶解,然后加入相應(yīng)質(zhì)量的環(huán)氧化硅氧烷(ES)、1 g 六次甲基四胺和咪唑(添加量為ES質(zhì)量的5%),室溫下攪拌1 h,使加入物質(zhì)在溶劑中充分溶解。隨后利用旋轉(zhuǎn)蒸發(fā)儀和真空干燥箱除去溶劑,得到黃色固體,即為ES和NR共混預聚體樹脂,記為NR-PES。其中1-NR-PES、2-NR-PES 和4-NR-PES 分別代表NR 與ES 的添加量的質(zhì)量比為1∶1、2∶1和4∶1。
將上述所得的共混預聚體樹脂在合適模具中以80℃+100℃+200℃的溫度固化,得到橙黃色透明樹脂樣條,具體固化溫度和時間由DSC 和流變學測試結(jié)果確定。
1.3 測試方法
采用DSC 214 polyma型號差示掃描量熱儀對樣品進行測試,研究其固化行為,具體測試條件如下:升溫范圍為30 ~300℃,升溫速率為5、10、15、20℃·min-1,氮氣氣流恒定為50 ml·min-1;采用MCR 302流變儀在等溫恒頻率流變和等溫變頻率流變兩種模式對NR-PES 樣品進行表征測試。等溫恒頻率流變是分別以80、100 和200℃為溫度點,在恒定剪切頻率(ω=1 rad·s-1)和應(yīng)變(γ=1%)的條件下進行測試,以研究樹脂儲能模量(G')、損耗模量(G″)與時間的變化關(guān)系。在等溫變頻率測試模式中,樣品在100℃恒溫度條件下,進行0.01~100 rad·s-1范圍內(nèi)多次變頻測試,掃描時間為每次20 min,重復多次掃描直至樣品的儲能模量值趨于穩(wěn)定;采用DMA Q800(TA Instrumental Company)動態(tài)熱機械分析儀對不同預聚程度NR-PES 樹脂樣條進行分析測試,研究預聚程度對樹脂模量和交聯(lián)密度的影響。具體測試條件如下:采用單懸臂梁夾具,測試溫度范圍為30~250℃;升溫速率為3℃·min-1,應(yīng)變?yōu)?%,頻率為1 Hz,樣品尺寸大小約為12 mm×3 mm×35 mm,并根據(jù)橡膠彈性理論[28],通過式(1)計算固化樹脂的交聯(lián)密度;采用TG209C 熱重分析儀對不同預聚程度NR-PES 體系固化后樹脂進行分析測試,研究其在氮氣條件下的熱穩(wěn)定性。具體測試條件如下:溫度測試范圍為30~800℃;升溫速率為10℃·min-1,氮氣氣體流量為50 ml·min-1;采用AVANCE ⅢHD 型號核磁共振波光譜測定樣品1H NMR,DMSO-d6 為溶劑,TMS 為內(nèi)標,掃描64次;根據(jù)標準ASTM D5045,利用萬能試驗機測定不同預聚程度NR-PES 固化樹脂樣條的斷裂韌性(KIC);根據(jù)標準GB∕T 2567—2021,測試固化樹脂的彎曲強度;利用MAIA3 LMH場發(fā)射掃描電子顯微鏡(SEM)對斷裂韌性測試樣條的斷面進行表征。

式中,Ve為交聯(lián)密度,mol·m-3;Er為Tg+40℃時的儲能模量,MPa。
2 實驗結(jié)果與討論
2.1 ES的結(jié)構(gòu)表征
合成的環(huán)氧化硅氧烷(ES)的FTIR 與1H NMR譜圖如圖2 所示。圖2(a)為兩種合成原料1,1,5,5-四甲基二苯基三硅氧烷和4-乙烯環(huán)氧環(huán)己烷及合成產(chǎn)物ES 的FTIR 譜圖,其中在842 cm-1和914 cm-1處的特征吸收峰屬于環(huán)氧基官能團,1081 cm-1和1131 cm-1是Si—O—Si 的伸縮振動吸收峰,且產(chǎn)物ES 在1635 cm-1處屬于—CH CH—以及2130 cm-1處屬于Si—H 的特征吸收峰完全消失,證明反應(yīng)完全。圖2(b)為ES的1H NMR譜圖,化學位移為2.954處出現(xiàn)單峰,屬于環(huán)氧基中的—CH—,化學位移在1.887和1.883處出現(xiàn)雙峰,屬于環(huán)己基中的—CH2—,化學位移為1.547 處出現(xiàn)的單峰,屬于環(huán)己基中的—CH—,化學位移為1.074和0.392處分別出現(xiàn)的雙峰,屬于硅氧基與環(huán)己基之間連接的—CH2—。如表1 所示,吸收峰的個數(shù)以及面積和每個吸收峰的化學位移都與合成的ES 結(jié)構(gòu)吻合,說明1,1,5,5-四甲基二苯基三硅氧烷和4-乙烯環(huán)氧環(huán)己烷在催化劑的作用下通過硅氫加成反應(yīng)成功合成了環(huán)氧化硅氧烷(ES)。
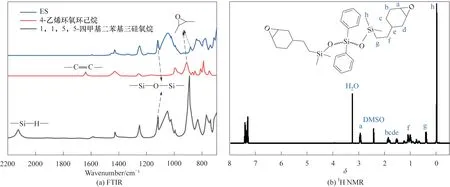
圖2 ES的FTIR和1H NMR譜圖Fig.2 FTIR and1H NMR spectra of ES
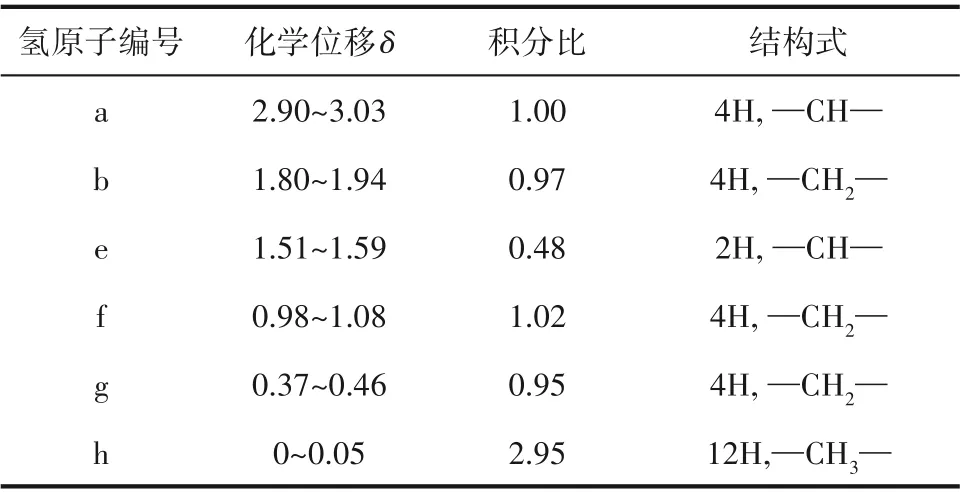
表1 ES的1H NMR譜圖分析Table 1 1H NMR spectrum analysis of ES
2.2 NR-PES的固化行為
通過非等溫DSC法研究了NH、1-NR-PES和2-NR-PES 的固化過程。從圖3(a)NR-PES 的DSC 曲線中可以明顯看出,ES中大量的活性環(huán)氧基的加入使得NR-PES 的固化窗口變寬,峰值溫度也向低溫方向移動。隨著PES 添加量的增加,NR-PES 的起始固化反應(yīng)溫度變低,固化窗口變寬。PES 添加量增大至50% 時,1-NR-PES 的固化窗口為53.4~201.0℃,放熱焓ΔH增大到220.33 J·g-1。大量活性較高的環(huán)氧基使得體系放熱焓增加(表2)。
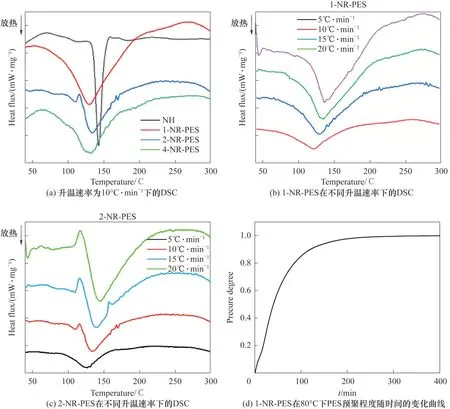
圖3 NR-PES的DSC曲線Fig.3 DSC curve of NR-PES system
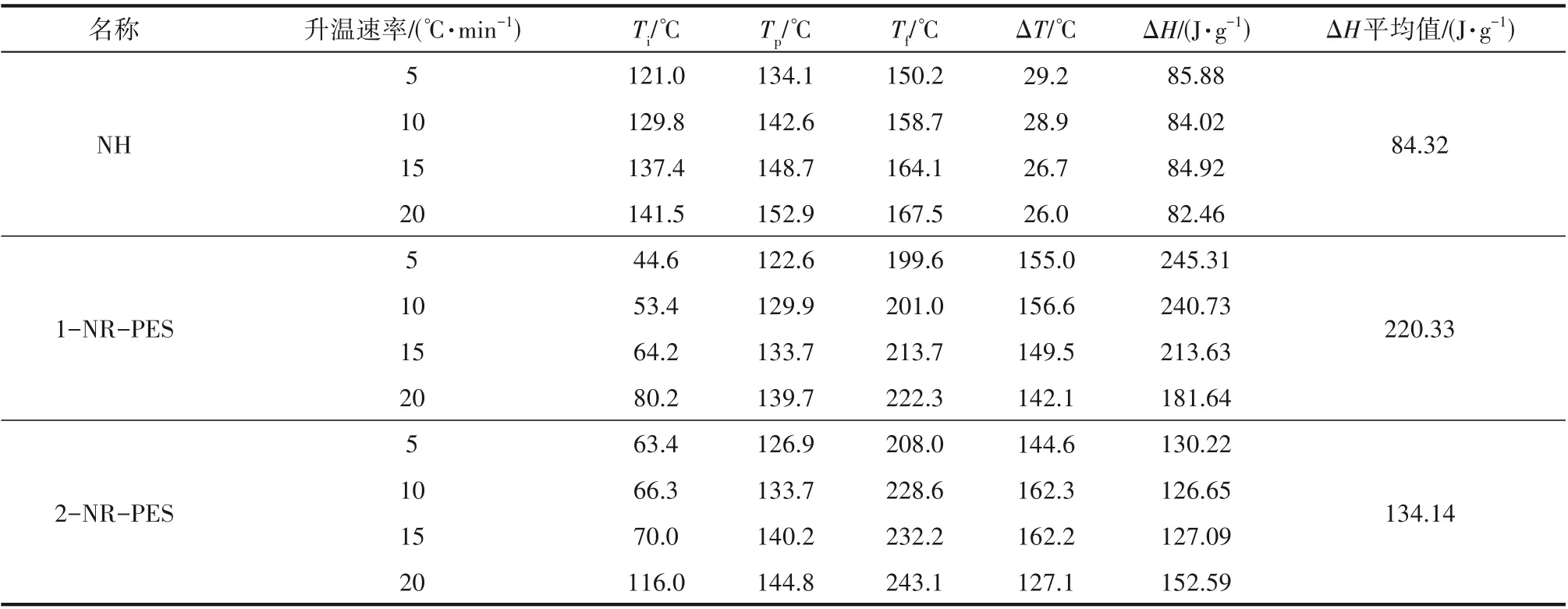
表2 非等溫DSC下的NH、1-NR-PES和2-NR-PES的固化特征參數(shù)Table 2 Curing characteristic parameters of NH,1-NR-PES and 2-NR-PES under non-isothermal DSC
此外,根據(jù)現(xiàn)有的研究可知,80℃下ES 中環(huán)氧基在咪唑作用下發(fā)生自聚反應(yīng)[29],形成不同鏈長的柔順性預聚體PES(圖4),而在100℃下體系中未反應(yīng)的環(huán)氧基可以與酚羥基發(fā)生共聚反應(yīng)[30],形成不同交聯(lián)結(jié)構(gòu)的改性樹脂。因此通過調(diào)控在80℃下的固化時間,在共混樹脂體系中可以產(chǎn)生不同預聚程度的PES,再經(jīng)過100℃的固化,使PES 與酚醛樹脂共聚形成交聯(lián)網(wǎng)絡(luò)。
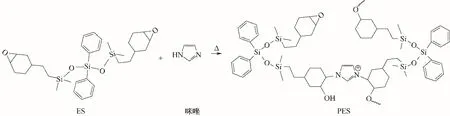
圖4 ES的自聚反應(yīng)示意圖Fig.4 Schematic diagram of ES self-polymerization reaction
在80℃下,共混樹脂主要發(fā)生ES 的自聚反應(yīng),其預聚程度α與時間的關(guān)系由式(2)通過儲能模量的變化計算得到[31],如圖3(d)所示。

此外,研究發(fā)現(xiàn),預聚體在固化過程中先達到玻璃態(tài)再轉(zhuǎn)入后固化階段,有利于構(gòu)建致密交聯(lián)的熱固性樹脂,并提升耐熱性能[32]。基于此,進一步分析NR-PES 體系固化過程中的物理狀態(tài)(凝膠態(tài)和玻璃態(tài)),通過改變固化條件來使其先達到玻璃態(tài)再轉(zhuǎn)入后固化階段,制備不同PES 預聚程度的NRPES 固化樹脂,以進一步探討不同預聚程度的PES對NR-PES交聯(lián)網(wǎng)絡(luò)、熱性能和力學性能的影響。
2.3 NR-PES的等溫流變學
通過等溫流變學對儲能模量(G')和損耗模量(G″)的追蹤,可以準確確定NR-PES 到達凝膠態(tài)和玻璃態(tài)的所用時間。圖5 是1-NR-PES 體系在80、100 和200℃下的模量隨時間變化的曲線。當儲能模量(G')和損耗模量(G″)相等時,即為體系的凝膠點。因此,1-NR-PES 體系在80℃和100℃條件下的凝膠時間分別為120 min 和70 min。圖5(b)是1-NR-PES 經(jīng)200℃后固化的模量隨時間變化曲線,可以看出,在120 min 后體系模量趨于穩(wěn)定,表明后固化反應(yīng)完全,三維網(wǎng)絡(luò)結(jié)構(gòu)趨于穩(wěn)定。
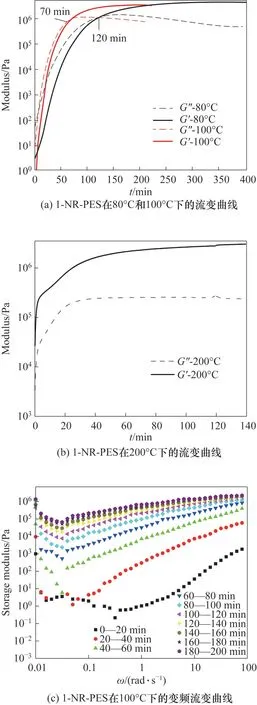
圖5 1-NR-PES的流變曲線Fig.5 Rheological curves of the 1-NR-PES system at different temperatures
利用流變測量變頻率下的儲能模量隨溫度的變化,可以確定NR-PES 到達玻璃態(tài)所需的時間。1-NR-PES 體系在100℃不同頻率(ω= 0.01~100 rad·s-1)下儲能模量隨時間的變化如圖5(c)所示:在t<140 min 時,體系的儲能模量在低頻率下隨時間變化而明顯增大,體系的交聯(lián)網(wǎng)絡(luò)結(jié)構(gòu)逐漸形成。而當t>140 min 時,體系的儲能模量較為穩(wěn)定,隨時間的變化較小,說明體系的交聯(lián)網(wǎng)絡(luò)結(jié)構(gòu)已經(jīng)趨于穩(wěn)定。因此,100℃下,140 min 可以作為1-NR-PES 體系到達玻璃態(tài)的時間。
基于NR-PES 非等溫DSC、等溫和變溫流變特性(圖5)的探討,NR-PES 的固化制度確定為:在80℃下到達設(shè)定的PES 預聚程度[固化時間確定見圖3(d)],然后在100℃下固化到達玻璃態(tài),再于200℃下達到完全固化。制備出的不同預聚程度PES改性NR樹脂固化物,記為1(或2)-NR-PES-X,X為PES 的預聚程度(0%、30%、60%、90%)。以1-NR-PES-0%固化制度為例,由于預聚程度為0%,未經(jīng)過80℃預聚階段,直接在100℃固化140 min 到達玻璃態(tài),然后升溫到200℃后固化120 min。而1-NR-PES-60%的固化制度則為80℃固化51 min 使PES 預聚程度達到60%,再于100℃下固化到達玻璃態(tài),在200℃固化120 min。
2.4 不同預聚程度NR-PES的動態(tài)熱力學性能
利用DMA 表征不同預聚程度1-NR-PES 和2-NR-PES 的熱力學性能,結(jié)果如圖6 和表3 所示。當初始組成為NR∶ES=2∶1 時,不同預聚程度2-NRPES 的熱力學性能差異較大。2-NR-PES-0%和2-NR-PES-30%的tanδ曲線類似,只表現(xiàn)出一個玻璃化轉(zhuǎn)變弛豫峰,Tg分別為121.13℃和122.02℃;初始儲能模量E'(40℃)和半峰寬也相差不大,說明兩者微相結(jié)構(gòu)的均一性相似。而當2-NR-PES 中PES 的預聚程度達到60%和90%時,tanδ曲線向低溫方向移動,并表現(xiàn)為不對稱雙峰,特別是2-NR-PES-90%表現(xiàn)出明顯雙峰,說明隨著樹脂中PES 預聚程度的增大,NR-PES 的微觀相結(jié)構(gòu)均一性下降,出現(xiàn)了一定程度的相分離。對2-NR-PES-60%和2-NR-PES-90%兩條tanδ曲線進行分峰擬合處理,如圖6(d)所示,得到兩個峰對應(yīng)的玻璃化轉(zhuǎn)變溫度Tg1和Tg2。不同預聚程度2-NR-PES 的橡膠態(tài)儲能模量E'(Tg+40℃)和交聯(lián)密度變化趨勢相同,隨著預聚程度的增加,先增大后降低。不同預聚程度2-NRPES 的交聯(lián)密度分別為3.20×10-3、4.05×10-3、4.38×10-3、4.18 × 10-3mol·m-3,2-NR-PES-60%的交聯(lián)密度達到最高。
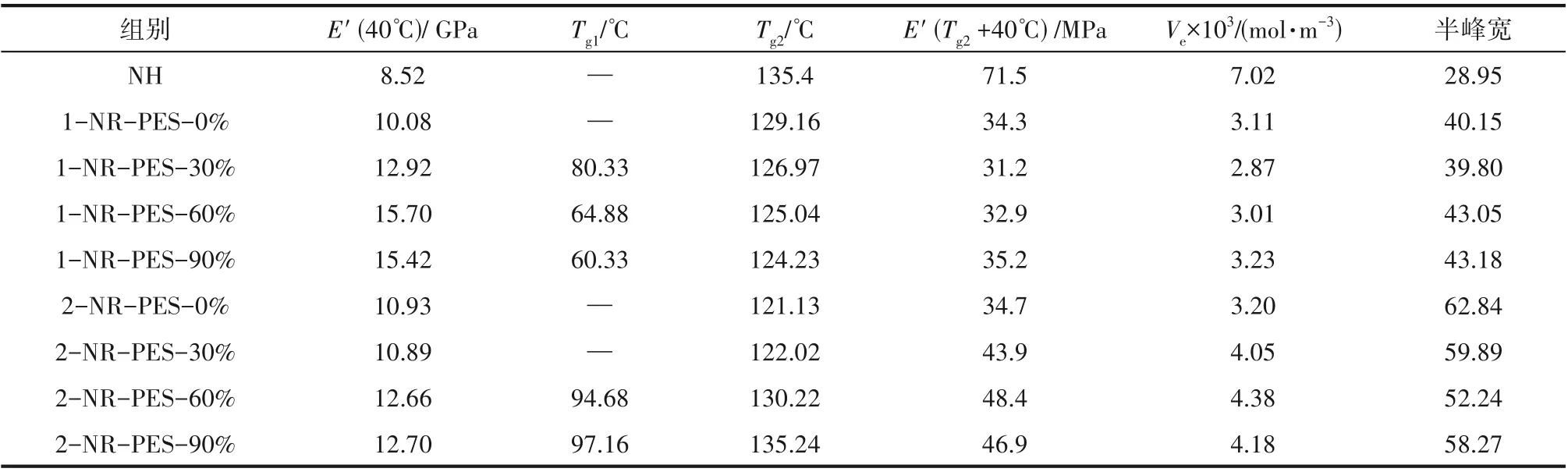
表3 NR-PES的動態(tài)熱-力學性能Table 3 Dynamic thermo-mechanical properties of NR-PES
而當初始組成為NR∶ES=1∶1時,不同預聚程度1-NR-PES 的熱力學性能較為接近。當1-NR-PES預聚程度為0%時,即NR 與ES 單體發(fā)生共固化,E'(40℃)為10.08 GPa,只表現(xiàn)出一個玻璃化轉(zhuǎn)變溫度Tg為129.16℃,且隨著預聚程度的增加,1-NR-PES的Tg逐漸降低,初始儲能模量E'(40℃)逐漸增大,預聚程度到達60%以上時基本保持在15 GPa 左右。1-NR-PES 的交聯(lián)密度隨著預聚程度的增加,先降低后升高。當PES 預聚程度為30%,交聯(lián)密度降到最低為2.87×10-3mol·m-3,隨后隨著預聚程度的增加交聯(lián)密度不斷增大。這是由于PES本身的醚鍵交聯(lián)具有高致密性的特點,PES聚合度提高時,能進一步增強樹脂的交聯(lián)密度。同時當PES預聚程度增加時[圖6(c)],1-NR-PES 的tanδ曲線在80℃出現(xiàn)了第二個弛豫峰,且隨著PES 預聚程度的增加而逐漸向低溫方向移動,對應(yīng)的峰值溫度分別為80.33、64.88和60.33℃。由于少量的未反應(yīng)ES 單體未連接到酚醛樹脂網(wǎng)絡(luò)結(jié)構(gòu)中造成相分離,ES 自聚程度增大,使得PES柔順性鏈段變長,玻璃化轉(zhuǎn)變溫度變低。
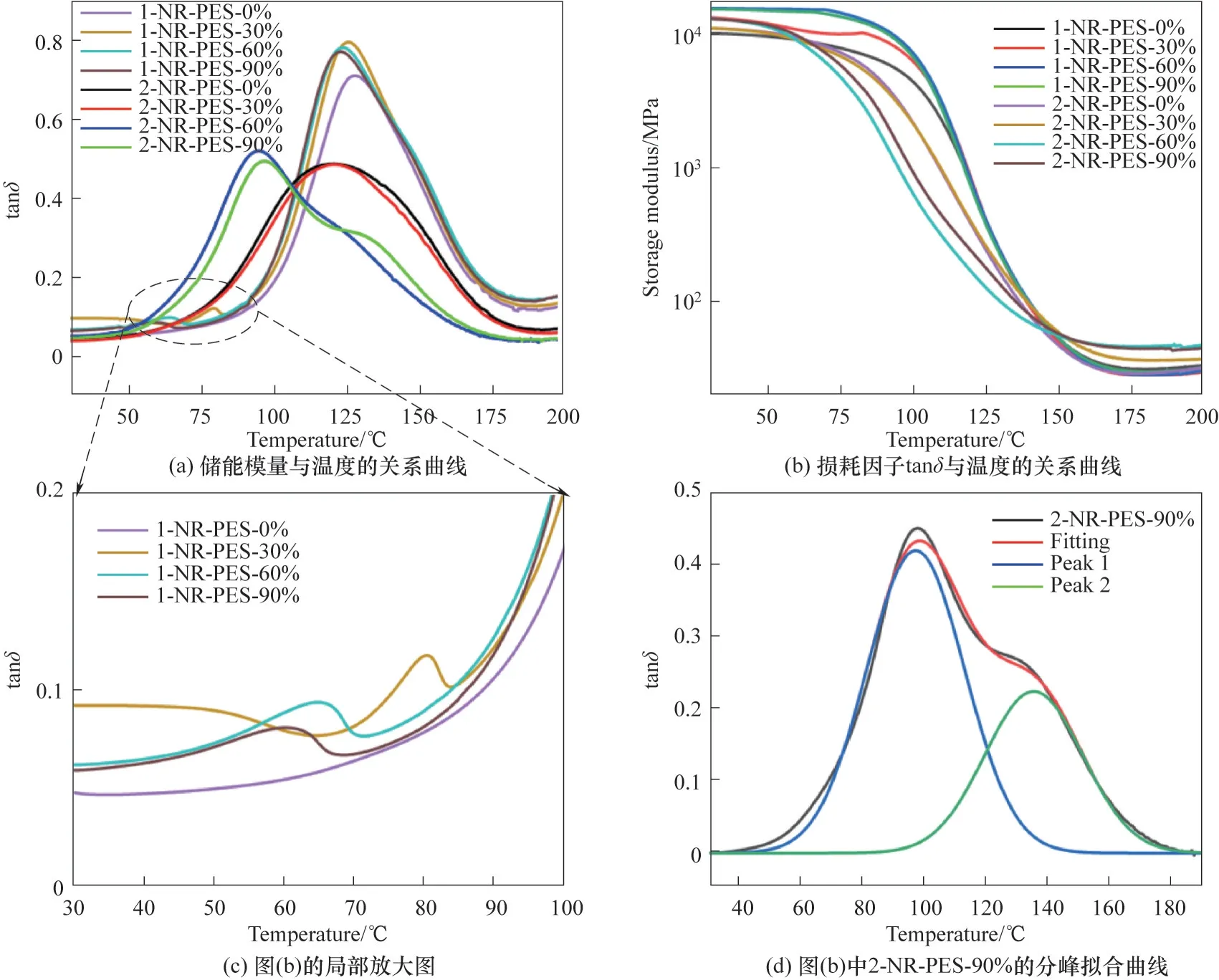
圖6 NR-PES的DMAFig.6 DMA curves of NR-PES systems with different prepolymerization degrees
總地來說,PES的添加量和預聚程度,會對NRPES 的交聯(lián)結(jié)構(gòu)產(chǎn)生顯著的影響。利用PES 鏈段的柔順性,將其引入到酚醛樹脂交聯(lián)網(wǎng)絡(luò)中,能有效降低酚醛樹脂過高的交聯(lián)密度,有利于降低酚醛樹脂因過高交聯(lián)密度導致的脆性。相同的預聚程度下,PES添加量較高時,對體系熱力學性能的改善效果越好,相比于2-NR-PES 體系,1-NR-PES 體系的熱-力學性能更加優(yōu)異。
2.5 不同預聚程度NR-PES的熱穩(wěn)定性
NR-PES 在氮氣條件下的TGA 譜圖,如圖7 和表4 所示。從圖7(b)中可以看到,不同預聚程度NR-PES的熱解過程與NH 相似,但整體的熱解溫度明顯向高溫方向移動。其中1-NR-PES體系的分解峰溫度(Td,max)在450℃左右,相比于NH 提高了約101℃,這是由于硅氧烷結(jié)構(gòu)的引入有效推遲了初始的熱分解溫度。但1-NR-PES 體系的熱分解速率DTG 相比于NH 有所升高,這是由于ES 上的脂肪族鏈段在高溫下容易裂解,導致熱解速率提高。不同預聚程度的1-NR-PES 的熱穩(wěn)定性變化不大,當預聚程度為30%時,1-NR-PES-30%具有相對較好的熱穩(wěn)定性,殘?zhí)柯蔆800℃提高到53.43%,而Dmax最低。
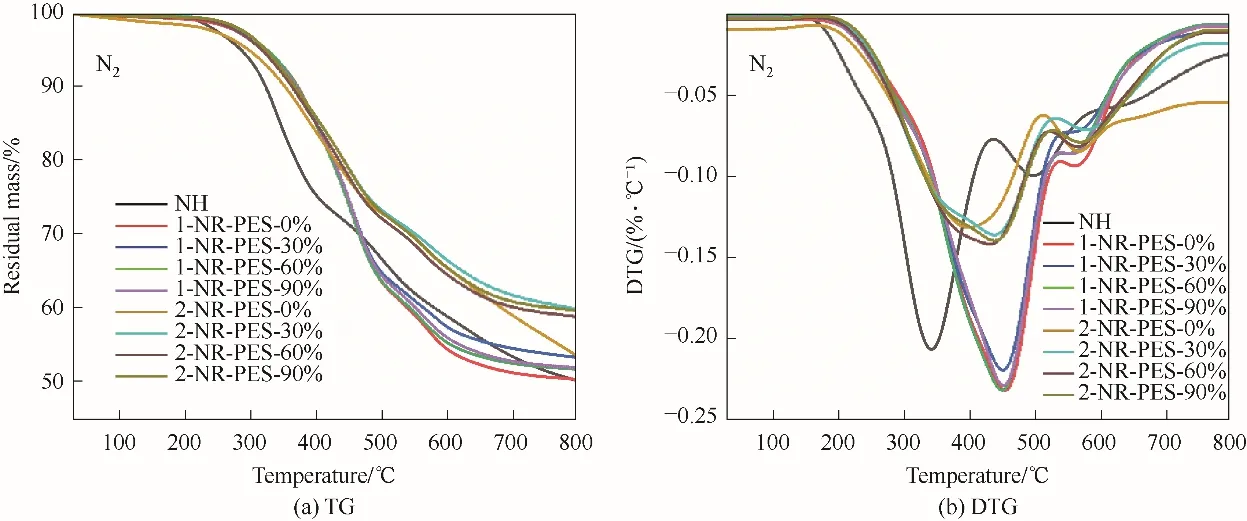
圖7 NR-PES在氮氣氣氛下的TG和DTG曲線Fig.7 TG and DTG curves of NR-PES under nitrogen atmosphere
相對于1-NR-PES 體系而言,不同預聚程度2-NR-PES 的熱穩(wěn)定性有明顯提升,特別是2-NRPES-30%,800℃下殘?zhí)柯蕿?9.92%,Dmax下降到-0.1358%·℃-1。2-NR-PES-0% 的T5%為300.5℃,Td,max為400.7℃,C800℃為53.64%。當PES 預聚程度增加到60%時,2-NR-PES-60%的T5%升高到320.0℃,Td,max提高至432.2℃,C800℃為58.93%。總體來說,不同預聚程度的PES與NR 共固化后,PES能有效增強改性樹脂的熱穩(wěn)定性。
2.6 不同預聚程度NR-PES 的力學性能和形貌表征
圖8 是NR-PES 體系的斷裂韌性(KIC)和彎曲強度。與NH 相比,NR-PES 體系的斷裂韌性得到顯著提高。隨著預聚程度的增加,1-NR-PES 體系的KIC先增加后逐漸降低,在PES 預聚程度為30%時達到最大值0.389 MPa·m1∕2之后,逐漸降低至0.216 MPa·m1∕2。當PES 預聚程度到達60%時,1-NR-PES 樹脂的彎曲強度最高為25.55 MPa,相比于NH 提高了32.2%。2-NR-PES 體系的KIC隨著PES 預聚程度的增大,從0.305 MPa·m1∕2小幅降低到0.252 MPa·m1∕2。PES 預聚程度為60%和90%時,2-NR-PES 的彎曲強度均提升。PES 預聚程度較低時,PES 與NR 基體樹脂相容性較好,其柔順性的鏈段對樹脂有明顯的增韌效果。結(jié)合圖9 樹脂斷面微觀形貌可知,未改性樹脂NH 的斷面呈現(xiàn)出光滑的脆性斷裂形貌,而不同預聚程度1-NR-PES 均表現(xiàn)為粗糙的鱗片狀態(tài),為韌性斷裂。而PES預聚程度過高時,其致密的交聯(lián)微相結(jié)構(gòu),會提升改性樹脂的強度。綜上所述,不同預聚程度的PES 均能有效增強樹脂的力學性能,且PES 的添加量和預聚程度對性能的影響較大。
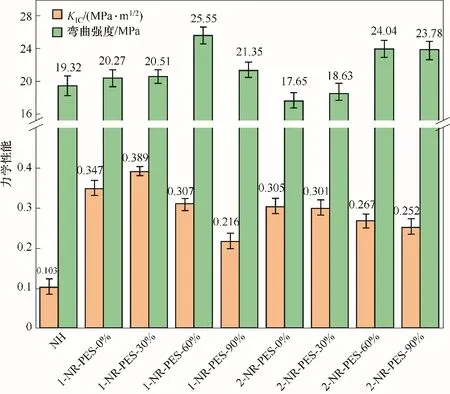
圖8 不同預聚程度NR-PES體系的力學性能Fig.8 Mechanical properties of NR-PES systems with different degrees of prepolymerization

圖9 1-NR-PES的微觀形貌Fig.9 Micromorphology of 1-NR-PES
3 結(jié) 論
本文利用硅氧烷的柔順性鏈段和Si—O—Si 耐熱結(jié)構(gòu)來改善酚醛樹脂的韌性和耐熱性,特別是明確了硅氧烷的預聚程度、鏈結(jié)構(gòu)、固化制度和固化物理狀態(tài)對改性酚醛樹脂結(jié)構(gòu)和性能的影響。具體結(jié)論如下。
(1)合成了一種脂環(huán)族環(huán)氧化硅氧烷(ES),通過FTIR與1H NMR對ES的結(jié)構(gòu)進行了表征。
(2)結(jié)合非等溫DSC 和等溫流變學分析,利用不同預聚程度的PES 改性熱塑性酚醛樹脂(NRPES)。通過控制NR-PES 在100℃下的固化時間,達到玻璃態(tài),再于200℃下完成后固化,獲得了具有不同網(wǎng)絡(luò)結(jié)構(gòu)的NR-PES。
(3)利用DMA、TGA、萬能力學試驗機、SEM 等手段研究了樹脂的組成和PES的預聚程度對其交聯(lián)網(wǎng)絡(luò)和力學性能的影響。當PES 添加量為50%(1-NR-PES),預聚程度為30%時,改性樹脂的熱穩(wěn)定性和斷裂韌性最好,C800℃為53.43%,KIC為0.389 MPa·m1∕2;而PES 預聚程度為60%時,初始儲能模量和彎曲強度達到最高,分別為15.70 GPa 和25.55 MPa。
猜你喜歡
新世紀智能(數(shù)學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國塑料(2016年12期)2016-06-15 20:30:07
中國塑料(2016年5期)2016-04-16 05:25:36
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
中國塑料(2015年4期)2015-10-14 01:09:19
現(xiàn)代企業(yè)(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
