高效液相色譜-串聯質譜法檢測水產品中 慶大霉素C組分殘留量
2022-11-21 09:24:54楊家鋒梁芹芹
現代食品 2022年20期
◎ 楊家鋒,鄭 丹,汪 杰,梁芹芹,蔣 海
(寧波市海洋與漁業研究院,浙江 寧波 315010)
慶大霉素在畜牧業及漁業養殖中應用廣泛,但同時也帶來了藥物殘留問題。慶大霉素及其殘留對人、畜均有明顯的毒副作用,長期攝入能誘發急性腎功能衰竭、中毒性耳聾、失語、癱瘓和休克等[2]。
慶大霉素的理化測定方法相當復雜,且靈敏度和通用性較差。因此應從多方面綜合考慮慶大霉素的殘留測定方法,本方法的主要改進措施有以下幾方面。①檢測過程盡量避免使用玻璃器具,可采用聚丙烯塑料管,進樣瓶亦采用塑料材質襯管等。②提取液為磷酸鹽緩沖加三氯乙酸,添加三氯乙酸既可有效去除蛋白質又能提高藥物的回收率。③采用MCX混合陽離子交換柱凈化濃縮提取液。④使用C18分離柱,流動相中加入五氟丙酸離子對試劑,有利于改善峰型和提高分離效果。⑤使用三重四極桿質譜配備電噴霧離子源,采用ESI+和MRM模式,提高檢測選擇性和靈敏度,克服了僅以保留時間作為定性依據的缺陷。
1 材料與方法
1.1 試劑
甲醇,乙腈:色譜純,美國西格瑪公司;七氟丁酸,五氟丙酸,三氟乙酸:色譜純,美國ACROS公司;磷酸氫二鈉:分析純,國產;0.01 mol·L-1磷酸鹽緩沖溶液:稱取1.36 g磷酸二氫鉀,量取30 mL三氯乙酸,用水溶解并定容至1 000 mL;磷酸二氫鉀:分析純,國產;氫氧化鈉:分析純,國產;1.0 mol·L-1氫氧化鈉溶液:稱取20.0 g氫氧化鈉,用水溶解并定容至500 mL;0.02 mol·L-1磷酸鹽溶液:稱取3.58 g十二水合磷酸氫二鈉,用水溶解并定容至500 mL,調整pH=7.0;氨水甲醇溶液:氨水+甲醇=10+90,v/v;1.0‰五氟丙酸溶液:稱取1.0 mL五氟丙酸,用水溶解并定容至1 000 mL;慶大霉素:純度≥98%, CAS No. 1405-41-0,Dr. Ehrenstorfer,美國;標準儲備溶液:1.0 mg·mL-1,稱取10.0 mg慶大霉素標準品,用水溶解并定容至10 mL,-18 ℃冷凍保存于塑料管中;標準使用液:準確吸取標準儲備液,用水稀釋配成 1 000 ng·mL-1溶液,現用現配;超純水:符合分析實驗室一級用水規格。
1.2 儀器設備
高效液湘色譜-串聯質譜儀,waters2695-QUTTRO MICRO,美國;分析天平,Sartorius BP310S,德國;均質器,FLUKO,上海;氮吹儀,HGC-24A;旋渦混合器,MS2 Minishaker,廣州;離心機,DL-8M,上海離心機研究所;真空旋轉蒸發儀,BüCHI R215瑞士;MCX陽離子固相交換柱,Waters,美國;固相萃取裝置:HENGAO T&D,HL-02D,巴西;移液槍:上海雷勃;超純水儀:美國Millipour公司。
1.3 樣品處理
1.3.1 制樣
取水產品可食部分,切成不大于0.5 cm×0.5 cm× 0.5 cm的小塊,充分勻漿,儲存于塑料瓶中,密封冷凍(-18 ℃)保藏,備用。
墻夼水庫總庫容 3.28億 m3,興利庫容0.85億 m3,死庫容 0.11億 m3,正常蓄水位98.50m。水庫工程防洪標準按百年一遇設計,萬年一遇校核。東、西庫設計洪水位分別為103.02m、103.16m,校核洪水位分別為106.50 m、106.57m。東西兩庫共設東庫溢洪閘1座,遭遇大洪水時,西庫水位高于東庫,洪水通過連通溝進入東庫,由東庫溢洪道泄出。
1.3.2 提取
稱取樣品(5.0±0.01)g置于50 mL離心管中,加入15 mL磷酸鹽緩沖溶液,振蕩10 min,設置 4 000 r·min-1轉速,離心15 min,上清液過濾后收集。殘渣再用15 mL磷酸鹽緩沖溶液重復提取1次, 4 000 r·min-1離心15 min后合并上清液濾液,用氫氧化鈉溶液調pH值為7.0~7.2,4 000 r·min-1離心 5 min后上清液過固相萃取柱。
1.3.3 凈化
MCX柱預處理:將MCX柱安裝到固相萃取裝置上,過提取液前先依次用5 mL甲醇,3 mL水,5 mL磷酸鹽緩沖溶液活化。提取液緩慢過已活化的MCX柱,再依次用2 mL磷酸鹽緩沖溶液和5 mL水洗雜質,待柱子干后以3 mL氨水甲醇溶液洗脫,收集洗脫液,氮氣吹干。精確量取1.0 mL流動相溶解殘留物,過 0.22 μm濾膜,待測。
1.4 標準工作溶液
吸取適量標準使用溶液(1.000 μg·mL-1)用水配制成序列濃度為10.0~1 000.0 ng·mL-1的標準工作溶液。
1.5 標準曲線制作
設定優化后儀器條件,將標準工作液上機測定,以慶大霉素各組分測得結果的峰面積或峰高為縱坐標,以標準工作液的濃度為橫坐標繪制標準曲線。
1.6 儀器條件
1.6.1 色譜條件
色譜柱:C18,100 mm × 2.1 mm,5 μm(i.d.),或其他性能相當的色譜柱;柱溫:室溫;進樣量: 30 μL;流動相:0.1%五氟丙酸酸溶液+乙腈=70+30。
1.6.2 質譜條件
離子化模式:大氣壓電噴霧離子源,ESI+;毛細管電壓:2.9 kV;錐孔電壓:25 V;脫溶劑氣流速:450 L·h-1;錐孔氣流速:50 L·h-1;離子傳輸毛細管溫度:350 ℃;離子源溫度:110 ℃;碰撞氣壓力:氬氣, 2.5 mTorr;掃描模式:多離子反應監測模式(MRM),選擇反應監測母離子、碎片離子和碰撞能量見表1。
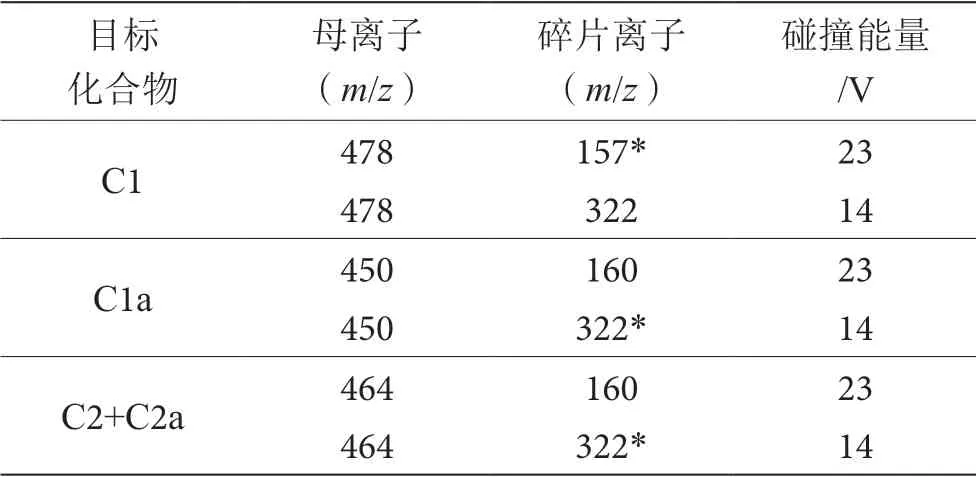
表1 選擇反應監測母離子、碎片離子和碰撞能量表
1.7 結果計算
樣品中慶大霉素殘留量按式(1)計算,計算結果需扣除空白值。

式中:Xi為樣品中慶大霉素C組分的含量,μg·kg-1;Ci為樣品制備液中慶大霉素各組分的濃度,ng·mL-1;V為最終定容體積,mL;m為樣品質量,g。
2 結果與分析
2.1 慶大霉素C組分的質譜特征
用水配制1 000 ng·mL-1的標準溶液用蠕動泵進樣方式直接導入ESI源,首先采用一級全掃描質譜方式,獲得待測物的準分子離子峰[M+H]+,然后對準分子離子峰及其碎片離子,采用Daughter scan質譜模式進行二級質譜分析,以獲取相應的多級質譜信息。
本實驗中的慶大霉素(圖1)由3個環組成,A環和C環為取代的氨基葡萄糖,B環為取代的脫氧鏈霉胺(中心單元)[3]。慶大霉素的結構中含有多個堿性中心,其裂解機制是由正電荷引發的i過程,失去的都是中性分子碎片,見圖2,表2。
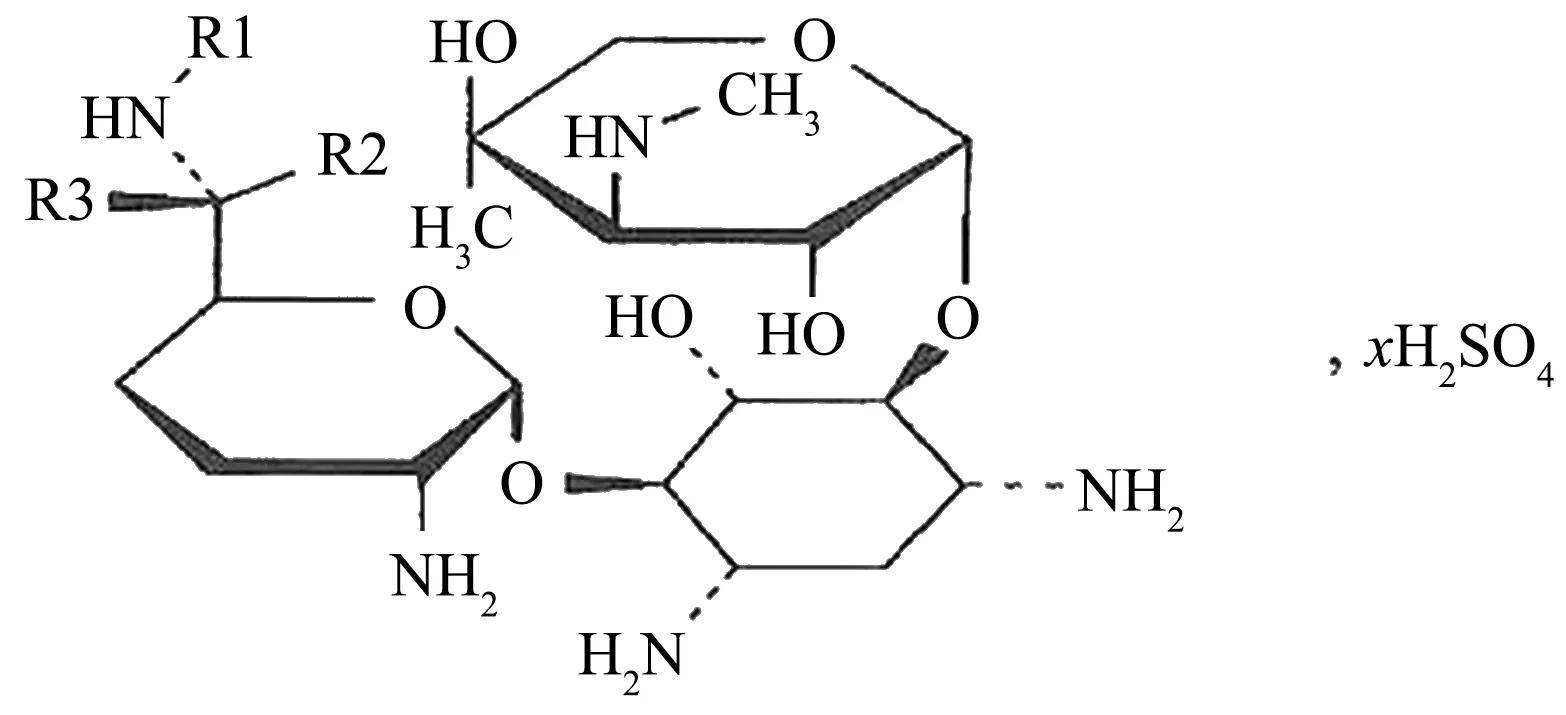
圖1 慶大霉素分子結構圖
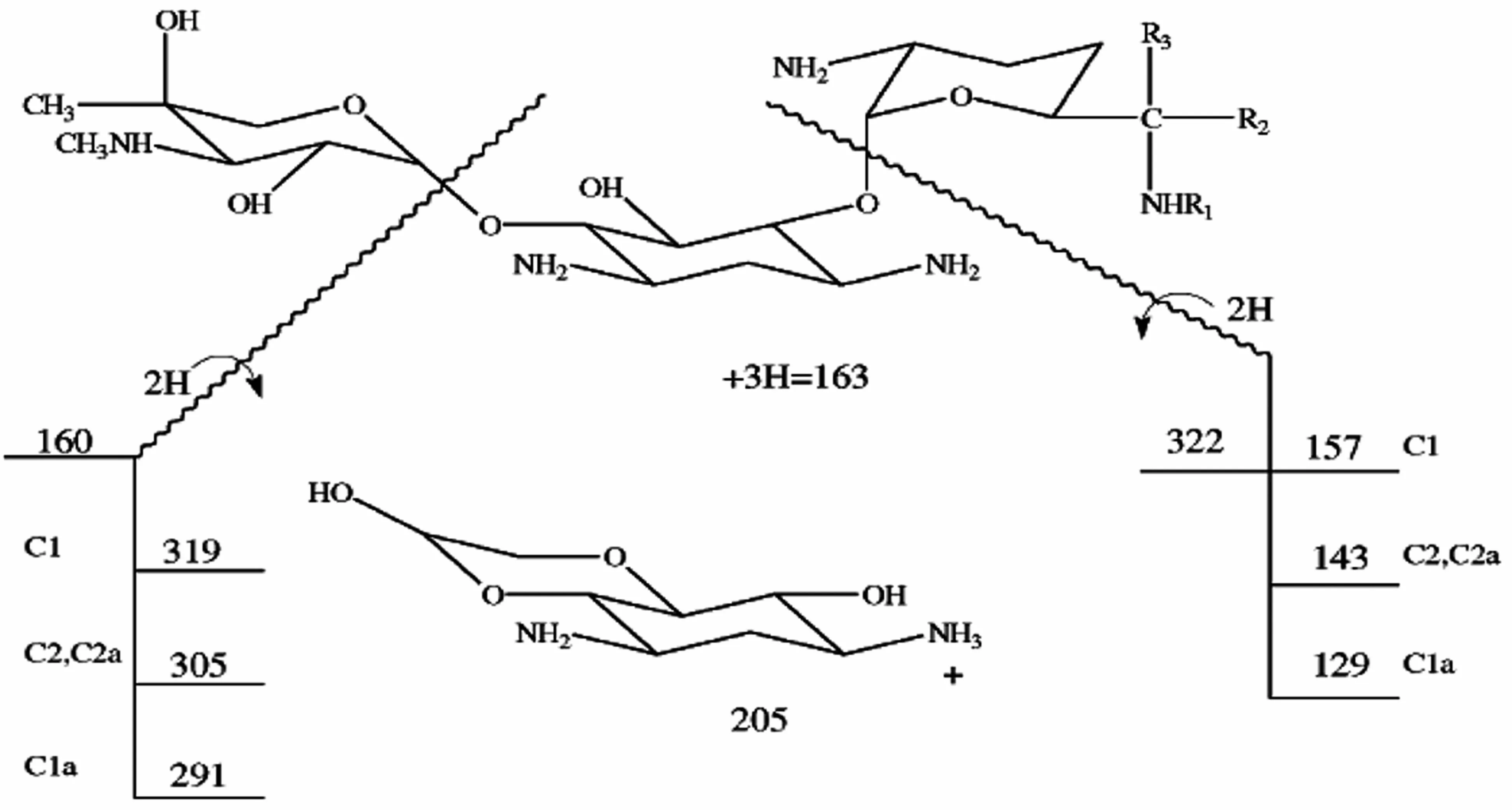
圖2 慶大霉素碎片離子裂解圖

表2 慶大霉素碎片離子裂解表
在一級全掃描質譜條件下,分別獲得4種結構相似的準分子離子峰m/z450,m/z464和m/z478(慶大霉素4種組分)(圖3)。為進一步了解該類抗生素的質譜斷裂規律,分別對各準分子離子峰進行多級質譜分析,獲得相應的碎片離子(圖4、圖5、圖6)。
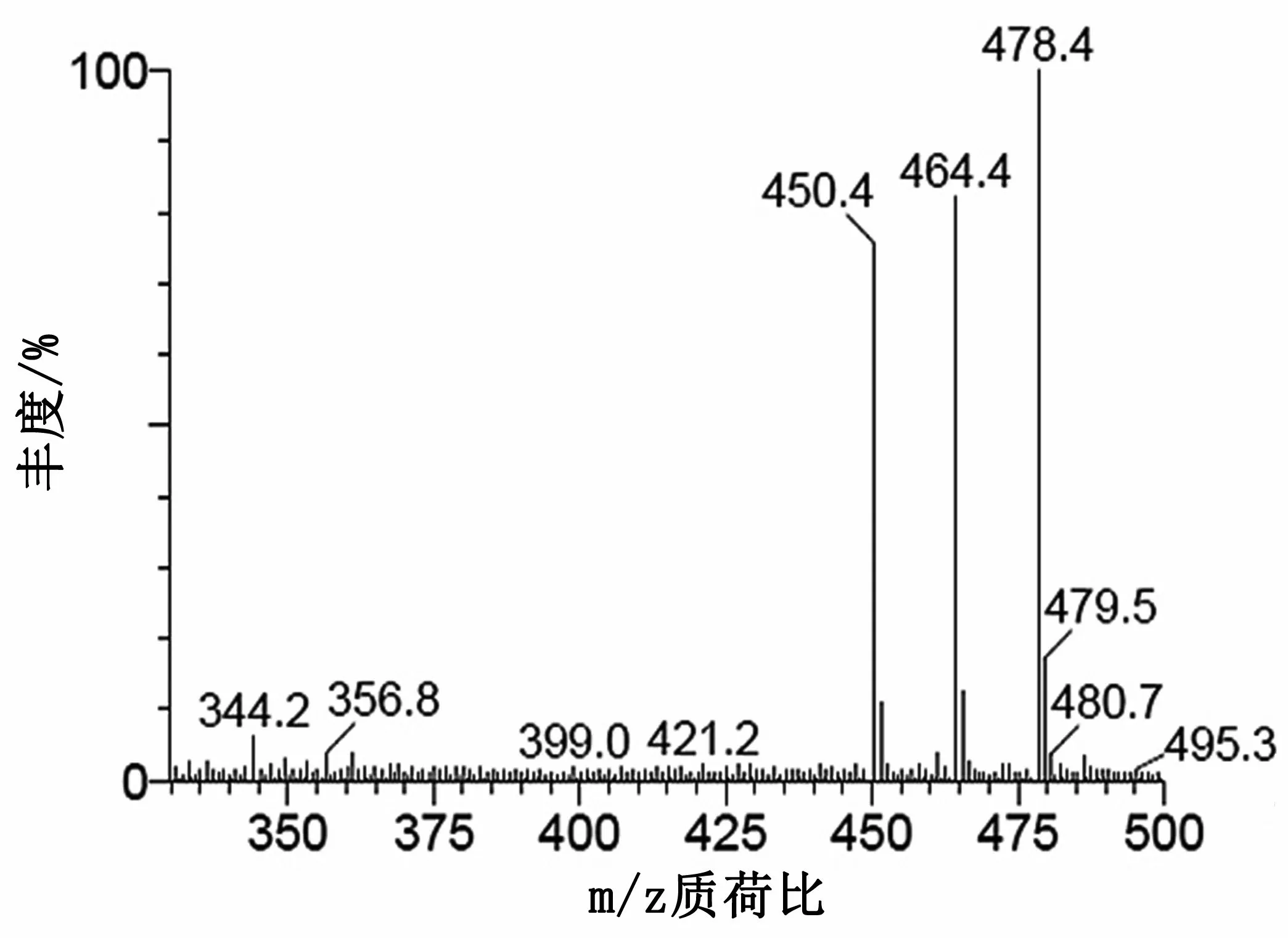
圖3 慶大霉素母離子質譜圖
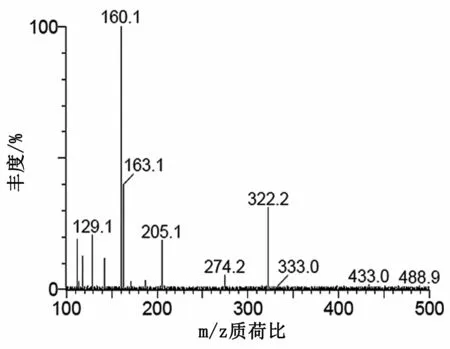
圖4 慶大霉素C1a子離子質譜圖
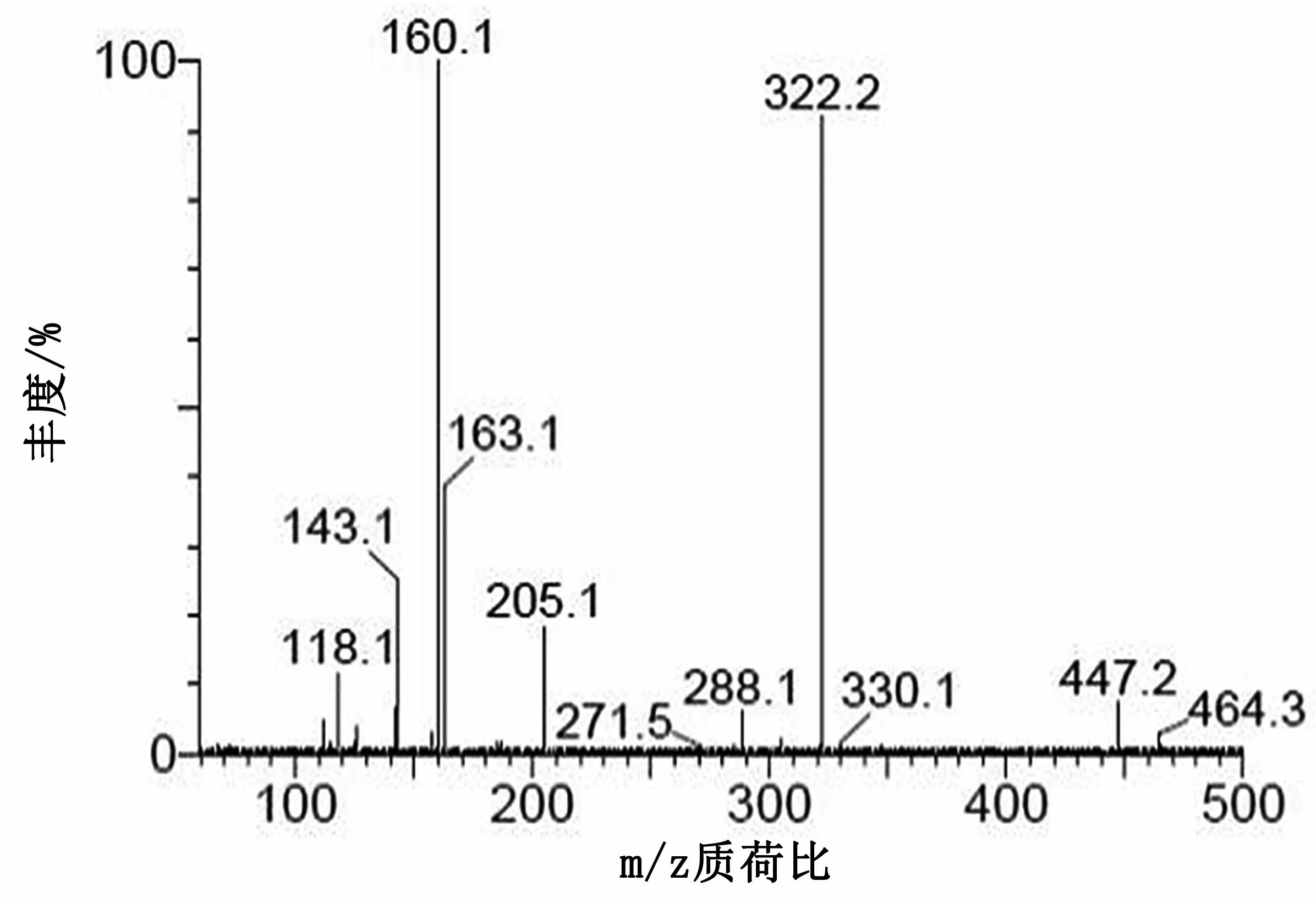
圖5 慶大霉素C2+C2a子離子質譜圖
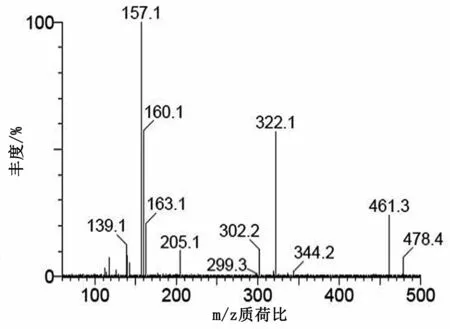
圖6 慶大霉素C1子離子質譜圖
在二級質譜分析中,慶大霉素C組分由4種(C1,C1a和C2+C2a)組成,對應3個準分子離子峰,分別選擇m/z478,m/z450和m/z464進行二級質譜分析,均形成同樣的碎片離子m/z322,進一步證明本實驗中的慶大霉素在二級質譜分析中首先脫去C環。加大碰撞能進行二級質譜分析,m/z322可生成m/z160(脫去B環)、m/z163(脫去A環)和m/z205等碎片離子。m/z160可進一步脫水分別生成m/z142碎片離子。m/z205推測是A環斷開,然后脫水形成的碎片離子。
2.2 質譜條件優化
1 μg·mL-1的慶大霉素標準溶液以蠕動泵方式進 樣,流速為20 μL·min-1,在ESI+模式下進行一級質譜圖掃描,以4種組分的[MH]+(C1,C1a和C2+C2a組成對應3個準分子離子峰分別為m/z478,m/z450和m/z464)峰靈敏度為依據,優化質譜調諧文件包括毛細管電壓、離子源溫度、錐孔電壓、脫溶劑氣等參數,見1.6.2。分別以m/z450,m/z478和m/z464作為母離子,對其子離子進行掃描,選取豐度最強的子離子m/z450>322,m/z478>322和m/z464>322為 定量離子,次強的子離子m/z450>160,m/z478>157和m/z464>160作為定性離子;以子離子強度為依據優化碰撞能量等的質譜條件見表1。
2.3 色譜條件的選擇
本實驗以三氟乙酸、五氟丙酸、七氟丁酸為離子對試劑進行試驗,濃度設0.5‰、1.0‰、1.5‰3個水平,以乙腈與離子對試劑溶液多組比例為流動相進行比較實驗。結果表明,用1.0‰五氟丙酸與乙腈比例為70∶30時,色譜峰的峰型、分離度和靈敏度都到達最優狀態,保留時間為2.91 min,標準溶液5 ng·mL-1時,定量子離子峰的信噪比大于15。隨著流動相中乙腈的濃度減少,目標物質保留時間延長。當乙腈含量為20%,保留時間為4.57 min,但靈敏度降低。以七氟丁酸為離子對試劑對慶大霉素的分離和提高色譜柱的保留有不錯的表現,在乙腈含量為20%,保留時間為7.21 min,但靈敏度比使用五氟丙酸時低。以三氟乙酸為離子對試劑,慶大霉素的色譜峰峰寬較大且有明顯峰拖尾。因此,選取1.0‰五氟丙酸+乙腈=70+30為流動相。
流動相中五氟丙酸濃度分別為0.5‰、1.0‰時,對保留時間及分離度的影響不大。流動相pH值在2.5~3.5內,容量因子改變不大,但pH值小于2.5或pH值大于4.0時,流動相容量因子急劇降低。在pH值為2.5~3.5時,五氟丙酸帶負電荷,而慶大霉素各組分分子帶正電荷,兩者形成離子對化合物,在反相色譜柱上被保留。1.0‰五氟丙酸的pH值在2.5~3.0,因此一般無需調節pH值。但當流動相pH值偏小時,慶大霉素各組分會出現雙峰現象。
在流動相為1.0‰五氟丙酸+乙腈=70+30時,比較了waters的Atlantis、Sunfire、Xterra、Symmetry以及資生堂的MGⅢ、CAPCELL-Pak混合柱等色譜柱對慶大霉素的出峰效果。結果表明,Atlantis C18的出峰效果最佳。
2.4 樣品處理條件優化
2.4.1 提取溶劑的確定
本方法以乙腈、二氯甲烷和乙酸乙酯有機溶劑以及磷酸鹽(KH2PO4)、高氯酸、三氯乙酸、三氟乙酸、氫氧化鈉作為提取溶劑進行回收率試驗。試驗結果表明用乙腈、二氯甲烷和乙酸乙酯有機溶劑提取試樣中慶大霉素藥物時,回收率很低,而且用乙酸乙酯提取時甚至沒有回收到目標物,該結果與慶大霉素的溶解性有關。氫氧化鈉溶液雖然能較好地提取試樣中慶大霉素,但在提取組織試樣時易形成乳膠狀態,不利于后續步驟的進行。2%~5%三氯乙酸或高氯酸既可有效脫蛋白質,也可以溶解慶大霉素提高回收率。三氟乙酸脫蛋白效果稍劣于三氯乙酸。因此,通過多組實驗比較后決定用磷酸鹽和三氯乙酸的混合溶液提取組織中慶大霉素,回收率高達89%~103%。DAVID等[4]采用30%的三氯乙酸提取血漿和尿中的慶大霉素,回收率為92%~107%,與本文結果較一致。
2.4.2 試樣的凈化
由于試樣中慶大霉素的提取劑為磷酸鹽和三氯乙酸的混合溶液,試樣提取液不易用旋轉蒸發的方式濃縮,固相萃取選擇吸附便是一種理想的濃縮凈化方法。本試驗以HLB、C18、MCX、SAX、WAX、PRS柱為凈化固相萃取柱,實驗結果表明C18、MCX、SCX對慶大霉素都有較好的保留,其中又以MCX結果最優,過柱回收率達97%以上,因此確定以MCX為本方法凈化固相萃取柱。過柱洗脫液為甲醇氨水溶液,在洗脫過程中第1 mL可將柱中95.5%的慶大霉素洗下,第2 mL又能洗下3.5%,因此確定洗脫液為3 mL。洗脫液氮吹至近干,準確量取1.0 mL流動相定容。ELIANGIRINGA等[5]使用CBA弱離子交換柱除去蛋白質后測定血清中的慶大霉素,回收率為78%~93%。
3 方法評價
3.1 回收率和精密度
以陰性大黃魚、南美白對蝦、梭子蟹為檢測試樣,每份5.00 g(稱準至0.01 g),分別添加慶大霉素,添加水平為分別為5 μg·kg-1、20 μg·kg-1、100 μg·kg-1。檢測結果表明,方法平均回收率在71.9%~101.0%,批內、批間變異系數均小于15%。本方法中認定的回收率為70%~110%。
3.2 線性關系和線性范圍
配制慶大霉素藥物標準的工作曲線濃度系列為 5 μg·L-1、10 μg·L-1、20 μg·L-1、50 μg·L-1、100 μg·L-1、200 μg·L-1、500 μg·L-1和800 μg·L-1,用液相色譜-串聯質譜分析,慶大霉素4種成分在5~800 μg·L-1具有很好的線性。慶大霉素4種成分標準溶液工作曲線見圖7、圖8、圖9,線性方程及定量限見表3。
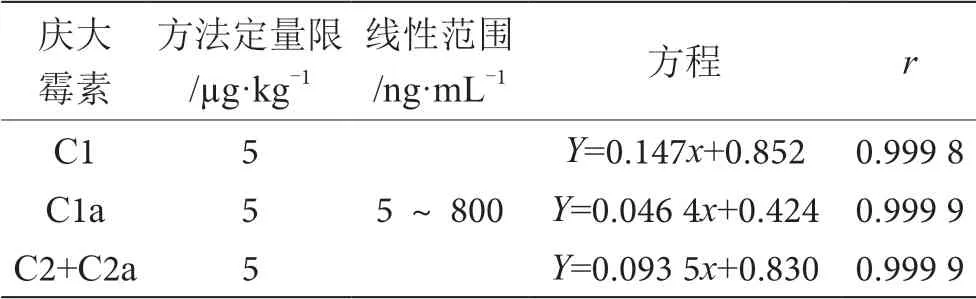
表3 慶大霉素標準溶液工作曲線及方法檢出限表
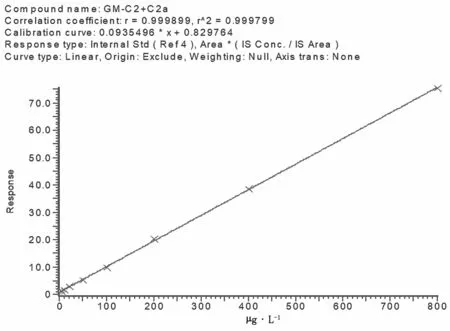
圖7 慶大霉素C2+C2a標準溶液工作曲線圖
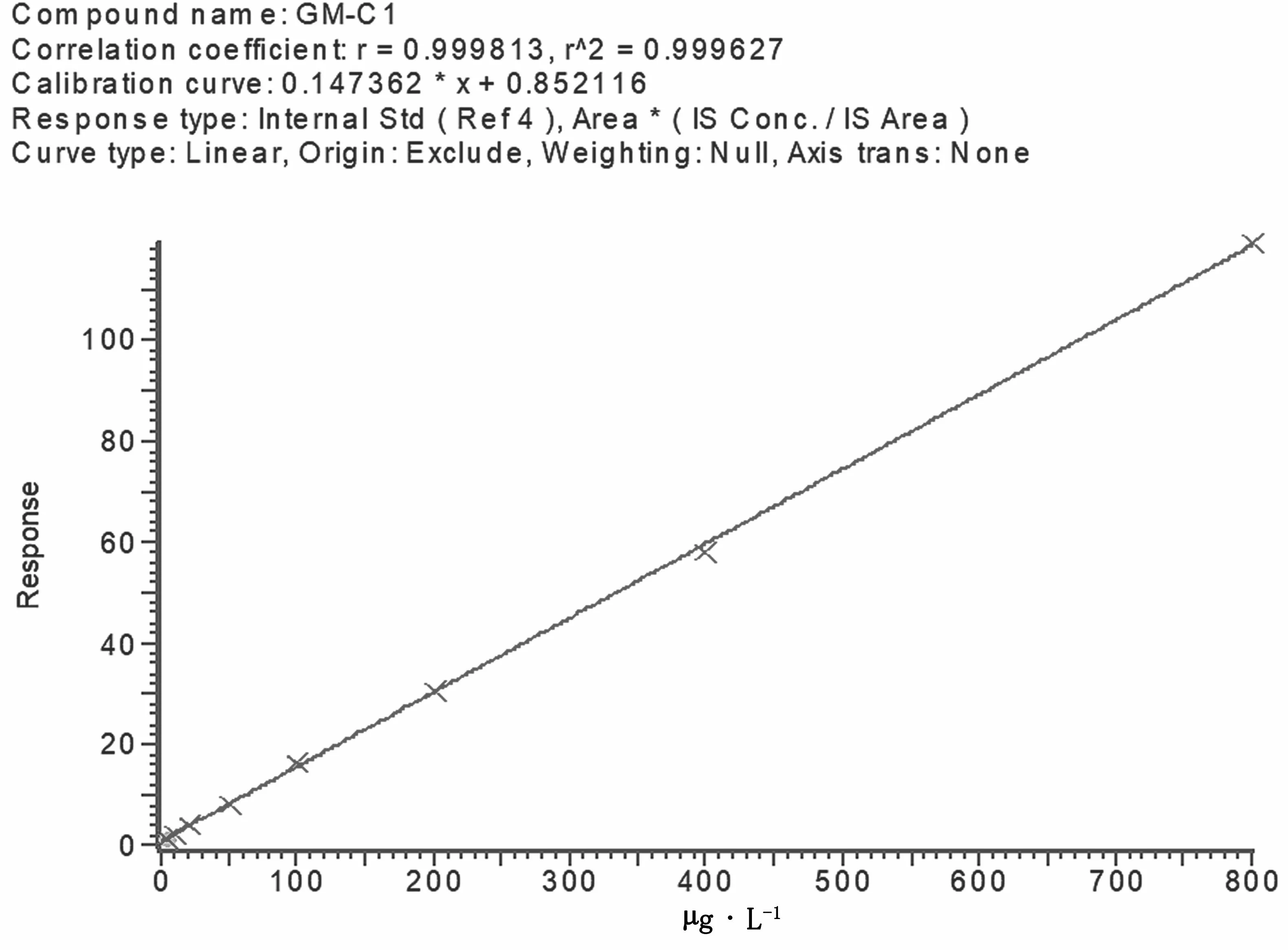
圖8 慶大霉素C1標準溶液工作曲線圖
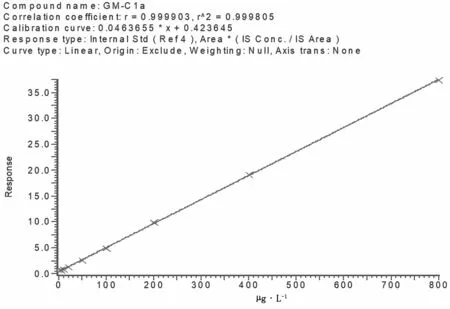
圖9 慶大霉素C1a標準溶液工作曲線圖
3.3 檢出限
對陰性大黃魚、南美白對蝦和梭子蟹分別以 5 μg·kg-1加標量進行加標回收試驗,試驗結果表明回收率都在71.9%以上,信噪比大于15。因此將5 μg·kg-1作為最小檢出濃度。
4 結論
本文構建了一種液相色譜-質譜聯用法檢測水產品中慶大霉素C組分殘留量的方法,實驗結果表明,慶大霉素4種C組分在0.005~0.800 μg·mL-1,線性相關系數良好,相關系數r>0.999 8;以5 μg·kg-1、20 μg·kg-1、100 μg·kg-13濃度水平進行加標回收實驗,回收率在71.9%~101.0%,RSD<15%;當加標量為5 μg·kg-1時4種C組分均具有明顯的響應峰,信噪比≥15。該方法適用于水產品中慶大霉素C組分殘留量的測定。
