QuEChERS-液相色譜-串聯質譜法測定 雞蛋中金剛烷胺和金剛乙胺殘留量
2022-11-21 09:25:02李巧蓮
現代食品 2022年20期
◎ 李巧蓮,劉 鑫,劉 瀛,林 洋,王 韻
(沈陽市食品藥品檢驗所,遼寧 沈陽 110136)
隨著人們生活水平的提高,雞蛋作為優質的高蛋白動物源食物,已成為餐桌上不可或缺的食物。因此,雞蛋中獸藥殘留問題引起人們高度關注。金剛烷胺和金剛乙胺是具有飽和三環葵胺結構的金剛烷胺類藥物,對流感病毒、帕金森氏綜合征等病癥有治療作用[1]。金剛烷胺和金剛乙胺可在家禽養殖過程中,用于預防禽流感和治療早期的禽流感,但是大量的金剛烷胺、金剛乙胺藥物殘留容易引起神經系統異常,對人體造成極大的危害[2-3]。因此,我國原農業部和美國食品藥品監督管理局在2005年和2006年先后頒布相關的公告,命令禁止在畜禽養殖環節中使用金剛烷胺、金剛乙胺類藥物[4-6]。但仍有部分養殖場違規使用金剛烷胺、金剛乙胺等類藥物,由此可見建立一種準確快速檢測金剛烷胺、金剛乙胺等類藥物殘留的方法具有重要意義。
目前測定金剛烷胺和金剛乙胺的方法主要有液相色譜法[7]、氣相色譜法[8]、酶聯免疫吸附法[9-10]及液相色譜-串聯質譜法[11]等。液相色譜法和氣相色譜法劣勢是靈敏度低、選擇性差、前處理過程煩瑣;酶聯免疫吸附法易出現假陽性;液相色譜-串聯質譜法因其高選擇性、高靈敏度、高通量等優勢,廣泛應用于多種農藥、獸藥殘留的檢測[12-15]。檢測雞蛋中金剛烷胺和金剛乙胺的方法常用的有國家標準《食品安全國家標準 動物性食品中金剛烷胺殘留量的測定 液相色譜-串聯質譜法》(GB 31660.5—2019)[16]和行業標準《出口動物組織中抗病毒類藥物殘留量的測定 液相色譜-質譜/質譜法》(SN/T 4253—2015)[17]。上述國家標準和行業標準是采用N-丙基乙二胺、固相萃取柱進行凈化。QuEChERS因其前處理過程快速、簡捷、有效等優勢,已經在食品藥品安全檢測領域中廣泛應用[18-21]。本研究用乙腈對雞蛋樣品進行提取,優化QuEChERS凈化技術,配比N-丙基乙二胺、十八烷基硅烷鍵合硅膠、石墨化碳黑(PSA-C18-GCB)3種吸附劑對樣品進行凈化,建立一種同時檢測雞蛋中金剛烷胺和金剛乙胺殘留的液相色譜-串聯質譜(Liquid Chromatography-Tandem Mass Spectrometry,LC-MS/MS)分析方法,以期為雞蛋中金剛烷胺和金剛乙胺殘留的分析和監管提供技術支持。
1 材料與方法
1.1 儀器與試劑
LCMS-8050液相色譜質譜聯用儀(日本島津公司);PTY-B620電子天平(感量0.01 g,華志電子科技有限公司);T10分散機(德國IKA公司);MS200多管渦旋混勻儀(杭州瑞誠儀器有限公司);H1750R離心機(長沙湘儀離心機儀器有限公司);C18色譜柱(150 mm×2.1 mm,3.5 μm,美國Waters 公司)。
標準物質金剛烷胺、鹽酸金剛乙胺、金剛烷胺-D15、鹽酸美金剛-D6(100 mg·L-1,上海安譜實驗科技股份有限公司);甲酸(分析純,國藥集團化學試劑有限公司);乙腈、甲醇(色譜純,美國Thermo Fisher Scientific公司);無水MgSO4、PSA、HC-C18、GCB(上海安譜實驗科技股份有限公司);實驗室用水為高純水。
樣品購自沈陽市超市。
1.2 實驗方法
1.2.1 標準溶液的配制
配制金剛烷胺、金剛乙胺混合標準中間溶液 (100 ng·mL-1):分別精密量取金剛烷胺、鹽酸金剛乙胺標準溶液100.0 μL,置于100 mL容量瓶中,并用甲醇定容至刻度,-18 ℃避光保存。
配制金剛烷胺-D15、鹽酸美金剛-D6混合同位素內標工作溶液(100 ng·mL-1):分別精密量取金剛烷胺-D15、鹽酸美金剛-D6標準溶液100.0 μL,置于100 mL容量瓶中,并用甲醇定容至刻度,-18 ℃避光保存。
1.2.2 液相色譜條件
C18色譜柱(2.1 mm×150 mm,3.5 μm);流動相:水相(A)為0.1%的甲酸水,有機相(B)為乙腈;洗脫梯度:0~1 min,5%B,1~4 min,5%B→95%B, 4~8 min,95%B,8~9 min,95%B→5%B, 9~11 min,5%B;流速:0.3 mL·min-1;柱溫:40 ℃;進樣體積:5 μL。
1.2.3 質譜條件
電噴霧正離子(ESI+)模式電離;多級反應監測模式(MRM);霧化氣的流量:3 L·min-1;干燥氣和加熱氣的流量:10 L·min-1;LD的溫度:250 ℃;接口的溫度:300 ℃;加熱塊的溫度:400 ℃。優化后定性離子對、定量離子對、碰撞能量(CE)參數見表1。
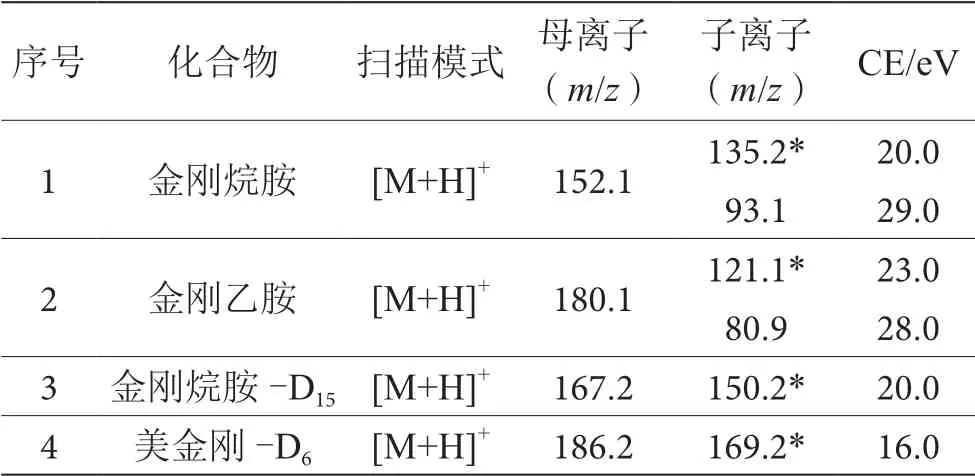
表1 質譜參數表
1.2.4 樣品前處理
稱取勻漿后的空白樣品5.00 g置于50 mL離心管中,加入同位素內標工作溶液(1.2.1)100.0 μL,加入10 mL乙腈,1 600 r·min-1渦旋提取5 min,在轉數為10 000 r·min-1的離心機中離心3 min,取上清液。將上述提取過程重復一次,合并混勻兩次上清液。向15 mL離心管中分別加入150 mg無水MgSO4和3種凈化劑PSA 100 mg、HC-C18100 mg 、GCB 25 mg,再加入4 mL 的上清液,1 600 r·min-1渦旋凈化5 min,在轉數為 10 000 r·min-1的離心機中離心3 min,取上清液,經 0.22 μm有機濾膜過濾后供液相色譜-串聯質譜儀分析。
2 結果與分析
2.1 質譜條件的優化
為獲得金剛烷胺和金剛乙胺最佳質譜參數,利用LCMS-8050液相色譜質譜聯用儀中的自動優化模塊,使用100 ng·mL-1的混合標準中間溶液(1.2.1)進行方法優化。利用前體離子搜索及產物離子搜索進行優化,得最優質譜條件,見表1。
2.2 色譜條件的優化
首先對乙腈-水和甲醇-水流動相體系進行考察,以乙腈為有機相時,可得到較好的色譜峰,色譜峰形尖銳對稱,且出峰附近無明顯的干擾峰,質譜響應值較高。進一步考察在水相中添加甲酸對質譜信號的影響,按0%、0.1%、0.2%、0.3%和0.5%改變添加甲酸的比例。實驗結果表明水相中添加0.1%甲酸時,化合物質譜信號明顯增加。但是隨著甲酸添加比例的增大,質譜信號明顯下降,如圖1。因為正離子掃描時,酸性條件下目標化合物易于獲取一個H+形成[M+H]+的分子離子峰,H+過多會抑制化合物的質譜響應。綜上,采用梯度洗脫程序,用0.1%甲酸水-乙腈作為流動相,對金剛烷胺、金剛乙胺進行分離。圖2為質譜和色譜條件優化后的MRM色譜圖。
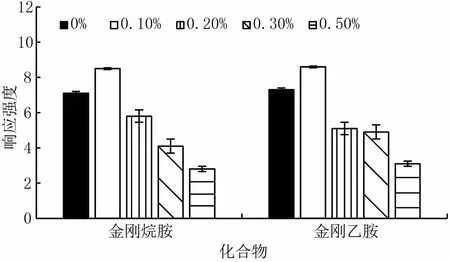
圖1 添加甲酸的比例對化合物離子化的影響圖(n=3)

圖2 MRM色譜圖(0.5 μg·L-1)
2.3 QuEChERS條件的優化
本研究優化了QuEChERS技術,優化脫水劑無水MgSO4和N-丙基乙二胺、十八烷基硅烷鍵合硅膠、石墨化碳黑(PSA-C18-GCB)3種吸附劑的組合用量。其中無水MgSO4用于吸附試樣中的少量水分,PSA用于吸附有機酸、脂肪酸、糖類,HC-C18用于吸附脂類等非極性干擾物,GCB能除去色素和固醇類物質的干擾。考察無水MgSO4、PSA、HC-C18和GCB用量對目標化合物回收率的影響。
稱取空白雞蛋基質5.00 g,添加一定量的混合標準中間液(1.2.1),使其質量濃度為0.5 μg·kg-1,按照1.2.4步驟前處理,取3組每組5個15 mL的離心管。①如圖3(a)所示,無水MgSO4的添加量(50 mg、100 mg、150 mg、200 mg和250 mg)對金剛烷胺、金剛乙胺回收率影響不明顯,實驗選擇添加量為150 mg。②添加150 mg無水MgSO4,改變PSA的添加量,如圖3(b)所示,當PSA添加量為150 mg時對金剛烷胺、金剛乙胺有明顯吸附,選取對化合物回收率最佳的添加量100 mg。③添加150 mg無水MgSO4和100 mg 的PSA,而改變HC-C18的添加量(25 mg、50 mg、100 mg、150 mg和200 mg),隨著HC-C18添加量的增加,如圖3(c)所示,化合物回收率明顯下降,HC-C18添加150 mg時吸附明顯,因此選擇HC-C18的添加量為100 mg。④添加不同量的GCB(15 mg、20 mg、 25 mg、50 mg和100 mg),再分別添加無水150 mg MgSO4、100 mg PSA和100 mg HC-C18,當添加25 mg的GCB時,金剛烷胺、金剛乙胺的回收率較高,如圖3(d)。

圖3 凈化填料用量對化合物回收率的影響圖(n=3)
綜上所述,選確無水MgSO4、PSA、HC-C18和GCB的添加量分別為150 mg、100 mg、100 mg和25 mg。
2.4 基質效應
基質效應(Matrix Effect,ME)普遍存在殘留分析的物質中,影響檢測結果的準確性,本研究采用基質標準曲線與甲醇為溶劑標準曲線線性方程斜率比進行計算評價。計算方法為 ME=(空白基質標曲斜率/溶劑標曲斜率-1)×100%,當ME為正值時為基質增強效應,當ME為負值時為基質減弱效應。弱基質效應為-20%~20%,基質干擾程度較低;中等程度基質效應為-50%~-20%和20%~50%;而ME小于-50%和大于50%為強基質效應,需采取措施補償基質效應[22]。表2結果表明,金剛烷胺、金剛乙胺為中等基質減弱效應。為消除基質干擾,提高檢驗結果的可靠性,應采用基質匹配校準曲線。

表2 基質效應、相關系數、檢出限和定量限表(n=3)
2.5 方法學驗證
2.5.1 檢出限、定量限和線性范圍
在空白基質中加入一定量的金剛烷胺、金剛乙胺標準溶液(1.2.1)使其濃度分別為0.125 μg·L-1、 0.250 μg·L-1、0.500 μg·L-1、1.250 μg·L-1和2.500 μg·L-1, 按1.2.4步驟進行操作,配制基質標準曲線。在 0.125~2.500 μg·L-1,相關系數r為0.999 87、0.999 90, 均大于0.99,線性關系良好。以信噪比S/N≥3和S/N≥10計算檢出限和定量限,得到的檢出限和定量限分別為0.5 μg·kg-1、2.0 μg·kg-1和2.0 μg·kg-1、5.0 μg·kg-1,如表2所示。
2.5.2 回收率和精密度
稱取空白基質5.00 g,添加一定量的混合標準中間液(1.2.1),使金剛烷胺質量濃度為0.5 μg·kg-1、 1.0 μg·kg-1和2.5 μg·kg-1、金 剛 乙 胺 質 量 濃 度 為 2.0 μg·kg-1、4.0 μg·kg-1和10.0 μg·kg-1,按1.2.4步 驟進行操作,對低、中、高3個濃度平行測定6次。如表3所示,平均回收率為93.43%~98.90%,相對標準偏差(RSD)為0.92%~4.14%。結果表明,該方法具有良好的準確度和較高的精密度。
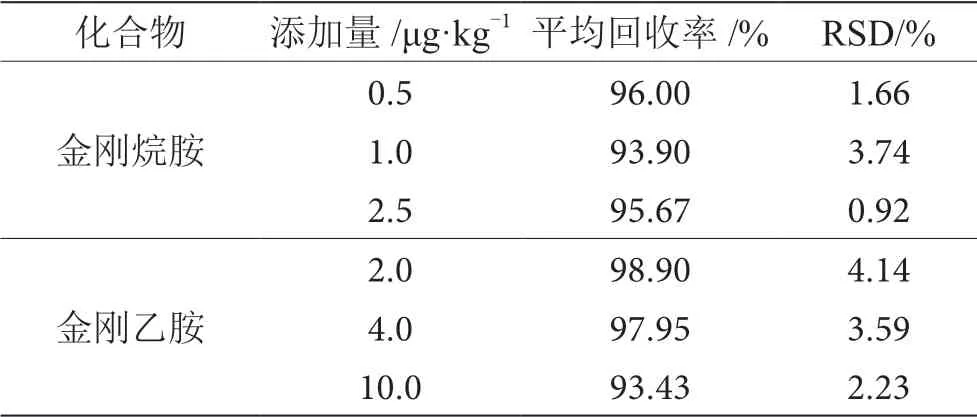
表3 回收率和精密度試驗結果表(n=6)
3 結論
本研究應用改進的QuEChERS技術結合液相色譜-串聯質譜法建立了雞蛋中金剛烷胺和金剛乙胺殘留量的檢測方法。采用基質標準曲線校準,同位素內標法定量。該方法前處理過程簡便、線性關系良好、準確度和精密度高,可為雞蛋中金剛烷胺和金剛乙胺殘留量的檢測提供參考依據。
