鼠原始生殖細胞報告基因系統的建立及驗證
2022-11-25 05:43:10黃書奇閻晗胡德慶
天津醫科大學學報 2022年6期
關鍵詞:小鼠
黃書奇,閻晗,胡德慶
(天津醫科大學基礎醫學院細胞生物學系,天津 300070)
胚胎干細胞(embryonic stem cells,ESCs)是一類具有無限增殖能力、多能性以及自我更新能力的細胞,可以分化成包括生殖細胞在內的多種細胞[1]。在小鼠早期胚胎發育過程中,胚胎基因組在受精卵形成后的2細胞時期開始激活。在8細胞時期,胚胎經歷緊束化形成桑椹胚,此時細胞開始出現極性,在胚內的一側形成一個充滿液體的腔,即囊胚腔,在囊胚腔的一側存在一個小的細胞團,即內細胞團(inner cell mass,ICM),ICM具有分化為成熟個體中全部細胞類型的潛能。隨著胚胎的繼續發育,ICM將快速增殖并進一步分化,逐步形成3個胚層以及相應的組織和器官[2-5]。哺乳動物胚胎植入子宮后,在BMP信號通路以及Wnt信號通路的共同作用下,近端外胚層細胞分化產生原始生殖細胞(primordial germ cells,PGCs)[6-9],鼠PGCs(mPGCs)呈堿性磷酸酶染色陽性,最早發現于原條后端,存在于胚胎發育的第6.25天(E6.25)。
基因轉錄調節因子Prdm1(PR/SET Domain 1)的蛋白序列中包含一個N端PR/SET結構域和5個近C端的C2H2鋅指結構域(鼠源Prdm1蛋白一級結構示意圖見圖1)。在早期胚胎發育過程中,Prdm1可以抑制體細胞形成相關信號通路的轉導,并致使部分外胚層細胞向生殖細胞譜系分化[10]。Dppa3(developmental pluripotency-associated 3)由母體效應基因編碼,主要在PGCs、植入前胚胎和其他多能干細胞中表達,其表達始于小鼠胚胎發育的第7.5天(E7.5),并持續表達至E15.5[11-12]。Dppa3主要包含N端結構域、SAP-like和Splicing-like結構域(包含核定位信號及出核信號)、C端結構域(鼠源Dppa3蛋白一級結構示意圖見圖1)。卵母細胞中Dppa3的缺失會導致囊胚數量減少、存活幼崽的數量下降[13]。Dppa3特異表達于PGCs和減數分裂前的生殖細胞,在PGCs特化過程中發揮了重要作用。
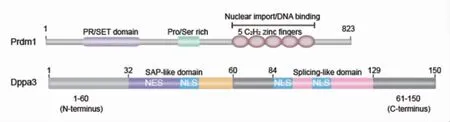
圖1 鼠源Prdm1、鼠源Dppa3蛋白一級結構示意圖Fig 1 Schematic diagram of the primary structure of mouse Prdm1 and Dppa3 proteins AB
研究表明,Dppa3與Prdm1在胚胎-胚外界面存在共定位[14-15]。并且,Prdm1和Dppa3為mPGCs的常用標志基因。本研究利用CRISPR基因編輯技術,在小鼠ESCs的Prdm1和Dppa3基因翻譯起始位點后分別插入eGFP和mCherry熒光蛋白編碼序列,構建了小鼠原始生殖細胞報告基因系統,對于研究小鼠早期胚胎發育過程具有重要意義。
1 材料與方法
1.1 材料
1.1.1 細胞系及質粒mESCs:V6.5購自美國模式培養物集存庫(American type culture collection,ATCC)。pSpCas9(BB)-2A-Puro(PX459)、pBluescript SK+質粒購自美國Addgene。
1.2 針對鼠Prdm1和Dppa3基因sgRNA的設計 利用網站(http://crispr.mit.edu)對鼠源Prdm1和Dppa3基因起始密碼子位置分別設計一對sgRNA,序列見表1,由生工生物工程(上海)股份有限公司合成。
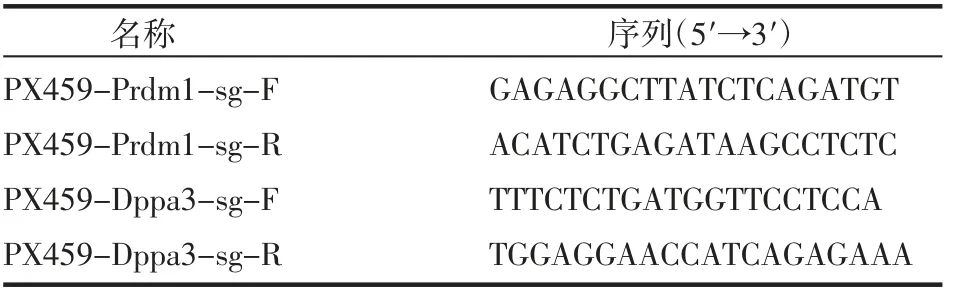
表1 sgRNA名稱及序列Tab 1 The name and sequences of sgRNA
1.3 Donor質粒的設計Donor質粒由pBluescript SK+載體骨架以及目的Donor片段構成,Donor片段主要由同源臂序列和要插入的目的片段構成。eGFP-Prdm1 Donor片段由6部分組成,第1部分為切割位點上游5′端500 bp同源臂,第2部分為綠色熒光標簽eGFP序列,第3部分為熒光標簽與目的基因Prdm1之間的Linker序列,第4部分為切割位點下游Prdm1 1號外顯子序列(去除ATG)及450 bp內含子序列,第5部分為FRT序列及Neomycin抗性基因序列,第6部分為3′端500 bp同源臂。mCherry-Dppa3 Donor片段設計原理同上。
1.4 mESC基因組DNA提取(1)收取細胞至1.5 mL EP管中,用200 μL Genomic Lysis Buffer重懸,加入終濃度為200 μg/mL的蛋白酶K,置于金屬浴55℃過夜。(2)每管加入200 μL異丙醇,顛倒混勻,置于-20℃靜置30 min,后4℃,13 500 r/min離心20 min。(3)棄掉上清,將沉淀用200 μL 75%乙醇洗2次,再用100%乙醇洗1次,棄掉上清,將沉淀晾干。(4)每管加入80 μL ddH2O,吹打混勻,將EP管置于55℃金屬浴1 h至沉淀徹底溶解。
1.5 PX459-sgRNA質粒及Donor質粒的構建
1.5.1 PX459-sgRNA質粒的構建(1)引物退火:利用降落PCR,使sgRNA與其互補鏈形成二聚體。體系:1 00 mmol/L濃度的sgRNA與其互補鏈各10 μL,ddH2O 30 μL;程序:95℃,5 min;-1℃/min,至25℃;4℃終止。(2)PX459載體線性化:用BbsI內切酶切割載體,體系:PX459載體1 μg;10×FD buffer 2 μL;FastDigestBbsI 0.5 μL;用ddH2O補至20 μL。程序:37℃,1 h。載體酶切產物在電泳后切膠,純化回收。(3)連接:體系:線性化PX459載體50 ng;雙鏈sgRNA 100 ng,10×T4 ligase buffer 2 μL;T4 ligase 1 μL;用ddH2O補至20 μL。室溫連接2 h。(4)轉化:將連接產物加入到100 μL DH5α感受態細胞中,冰上靜置30 min;42℃水浴熱激30 s;冰上靜置2 min,加入600 μL LB培養基,37℃,180 r/min搖菌45 min,轉化產物涂于Amp抗性的平板,37℃過夜培養,后挑取單克隆菌落,送公司進行測序。
1.5.2 Donor質粒的構建(1)pBluescript SK+載體線性化:用EcoR1和BamH1內切酶切割載體,體系:pBluescript SK+載體1 μg;10×FD buffer 2 μL;Fast-Digest EcoR1和BamH1內切酶各0.5 μL;用ddH2O補至20 μL。程序:37℃,1 h。載體酶切產物在電泳后切膠,純化回收。(2)eGFP-Prdm1和mCherry-Dppa3 Donor片段擴增:以mESC基因組DNA或質粒為模板,利用PCR擴出Donor片段。體系:5xHF Buffer 10 μL;10 mmol/L dNTP 1 μL;10 μmol/L上下游引物各1 μL;DMSO 1.5 μL;高保真DNA聚合酶0.5 μL;模板:若用質粒為模板,則加入10 ng,若用基因組DNA為模板,則加入250 ng;用ddH2O補至50 μL。程序:98℃預變性3 min;98℃變性30 s,52℃退火20 s,72℃延伸,延伸時間根據片段長度確定,本實驗中所使用的高保真DNA聚合酶延伸速率為30 s/kb,擴增循環數為32~35個循環;72℃延伸10 min。(3)連接:體系:線性化pBluescript SK+載體50 ng;Donor片段100 ng;T5 Mix 4 μL,用ddH2O補至20 μL。程序:30℃,40 min。(4)將連接產物轉化DH5α感受態中,37℃過夜培養,后挑取單克隆菌落進行菌落PCR鑒定,并送公司進行測序。
1.6 細胞培養及轉染mESCs在2i+LIF條件下培養,培養基成分為DMEM、15%FBS、0.1 mmol/L非必需氨基酸、2 mmol/L L-glutamine、1 mmol/L PD、3 mmol/L CHIR、1 000 U/mL LIF。將12孔板用0.1%Gelatin包被30 min,每個孔接種1×105個細胞,每兩天傳代一次。提取px459-sgRNA、pBluescript-eGFP-Prdm1和pBluescript-mCherry-Dppa3 Donor質粒。將px459-sgRNA和線性化pBluescripteGFP-mPrdm1 Donor質粒各2 μg轉染至2×106個細胞中。
1.7 單克隆細胞的篩選及鑒定 轉染24 h后更換培養基并加入1.5 μg/mL Puromycin培養48 h,后更換培養基并加入400 μg/mL Neomycin培養1周。挑取單克隆細胞至24孔板中培養,提取基因組DNA進行PCR鑒定。選取陽性細胞,將2 μg pCAGGSFLPe質粒轉染至2×106個細胞中,轉染24 h后更換培養基并加入1.5 μg/mL Puromycin培養48 h后撤藥,繼續培養1周后挑取單克隆細胞至24孔板中培養,提取基因組DNA進行PCR鑒定。
1.8 從mESCs向mPGCs的定向誘導 在體外誘導過程中,由mESCs向mPGCs的定向誘導主要由兩個步驟組成,第一步是mESCs在ActivinA和bFGF的作用下形成EpiLCs(epiblast-like cells),第二步是在BMP4、BMP8a、SCF、EGF、LIF的作用下由EpiLCs誘導形成PGCLCs(primordial germ cell-like cells)。
EpiLCs誘導:培養基成分為N2B27、1% KSR(Knockout serum replacement)、20 ng/mL ActivinA、12 ng/mL bFGF。將12孔板用0.1% Gelatin包被30 min,每個孔接種1×105個mESCs細胞,培養2 d,每天更換新鮮培養基,得到EpiLCs。
PGCLCs誘導:將2×103個EpiLCs接種到低吸附96孔板的一個孔中,PGCLCs培養基成分為GMEM、15%KSR、0.1 mmol/L非必需氨基酸、2 mmol/L L-glutamine、1 mmol/L sodium pyruvate、55 nmol/L 2-Mercaptoethanol、1 000 U LIF、500 ng/mL BMP4、500 ng/mL BMP8a、100 ng/mL SCF、50 ng/mL EGF。培養4 d后收集細胞,用流式細胞儀檢測熒光信號。
2 結果
2.1 CRISPR/Cas9基因敲入系統的構建
2.1.1 PX459-sgRNA質粒的構建結果 對PX459-eGFP-Prdm1-sgRNA和PX459-mCherry-Dppa3-sgRNA重組質粒進行測序,序列比對,編碼序列插入的位置和方向均正確,證明質粒構建成功(測序結果見圖2)。
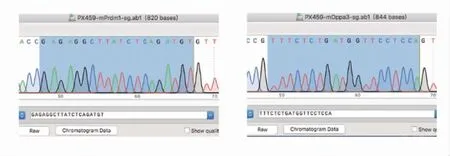
圖2 sgRNA測序結果Fig 2 The sequencing results of sgRNA
2.1.2 Donor質粒的構建結果eGFP-Prdm1 Donor片段由6部分組成,第1部分為切割位點上游5′端500 bp同源臂,第2部分為綠色熒光蛋白eGFP編碼序列,第3部分為eGFP編碼序列與目的基因Prdm1之間的Linker序列,Linker序列為5個連續的甘氨酸,第4部分為切割位點下游Prdm1 1號外顯子序列(去除ATG)及450 bp內含子序列,第5部分為FRT序列及Neomycin抗性基因序列,第6部分為3′端500 bp同源臂(圖3A)。mCherry-Dppa3 Donor片段同樣由6部分組成,第1部分為切割位點上游5′端500 bp同源臂,第2部分為紅色熒光標簽mCherry序列,第3部分為熒光標簽與目的基因Dppa3之間的Linker序列,第4部分為切割位點下游Dppa3 1號外顯子序列(去除ATG)及450 bp內含子序列,第5部分為FRT序列及Neomycin抗性編碼序列,第6部分為3′端500 bp同源臂(圖3B)。以mESCs基因組DNA為模板PCR擴增出Donor片段的第1、4、6部分,從含有相應序列的質粒上PCR擴增出第2、5部分,第3部分Linker序列為公司合成,根據DNA同源重組的原理使用T5酶將這些片段克隆到pBluescript SK+載體上,經公司測序,序列插入的位置和方向均正確,Donor質粒構建成功。
2.2 鼠Prdm1基因翻譯起始位點后插入eGFP編碼序列的細胞株鑒定 引物設計方案見圖3A。提取不同單克隆細胞的基因組DNA進行PCR,首先通過引物P3和P4從37個單克隆中鑒定出2個陽性克隆,以野生型(WT)細胞為對照,4號和37號克隆在750 bp位置出現條帶,說明這2個克隆均成功插入eGFP編碼序列。接下來通過引物P1和P2判斷這2個陽性克隆是純合子或是雜合子,其中WT在250 bp處出現條帶,4號克隆只在1 kb處出現條帶,為純合子;37號克隆分別在250 bp和1 kb處出現條帶,為雜合子。為了進一步確認eGFP編碼序列是否正確插入到鼠Prdm1基因翻譯起始位點后,從5′同源臂上游選取上游引物P7,在eGFP編碼片段上選取下游引物P8,PCR結果顯示,4號克隆和37號克隆均在700 bp位置出現條帶,說明eGFP編碼序列正確插入到鼠Prdm1基因翻譯起始位點后。
為了提高基因編輯的效率,在Prdm1的1號和2號外顯子之間的內含子上插入了Neomycin抗性編碼序列,其表達產物可以給細胞提供復制壓力,在使用Neomycin進行篩選時,細胞為了存活,會產生更多的同源重組,同時在Neomycin抗性編碼片段兩端插入了同向的FRT位點,外源表達的重組酶Flippase可以切除兩個方向相同的FRT位點之間的序列,從而達到去除Neomycin抗性編碼片段的目的。挑選4號和37號陽性克隆,轉入pCAGGS-FLPe質粒后用藥物進行篩選,提取不同細胞克隆的基因組DNA進行PCR,引物設計方案見圖3A。用引物P5和P6進行PCR,成功去除Neomycin抗性編碼片段的克隆無法擴出300 bp的條帶(圖4A)。
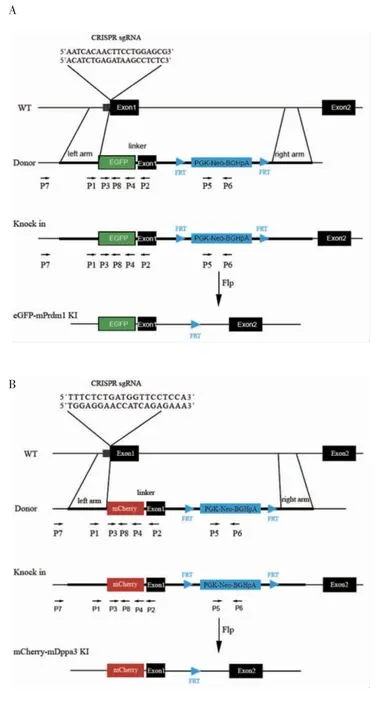
圖3 CRISPR介導的基因敲入策略模式圖Fig 3 Schematic diagram of CRISPR-mediated gene knock-in strategy
2.3 鼠Dppa3基因翻譯起始位點后插入mCherry編碼序列的細胞株鑒定 分別向上述4號和37號細胞克隆轉染PX459-mCherry-Dppa3-sgRNA和mCherry-Dppa3-Donor質粒,進行抗性篩選后挑取單克隆并提取基因組DNA進行鑒定,基因型鑒定引物設計方案見圖3B。挑取4號克隆中的純合子以及37號克隆中的雜合子,轉染pCAGGS-FLPe質粒去除Neomycin抗性基因片段。最后得到4-14號克隆為eGFP-Prdm1/mCherry-Dppa3雙純合子細胞,37-8號克隆為eGFP-Prdm1/mCherry-Dppa3雙雜合子細胞(圖4B)。
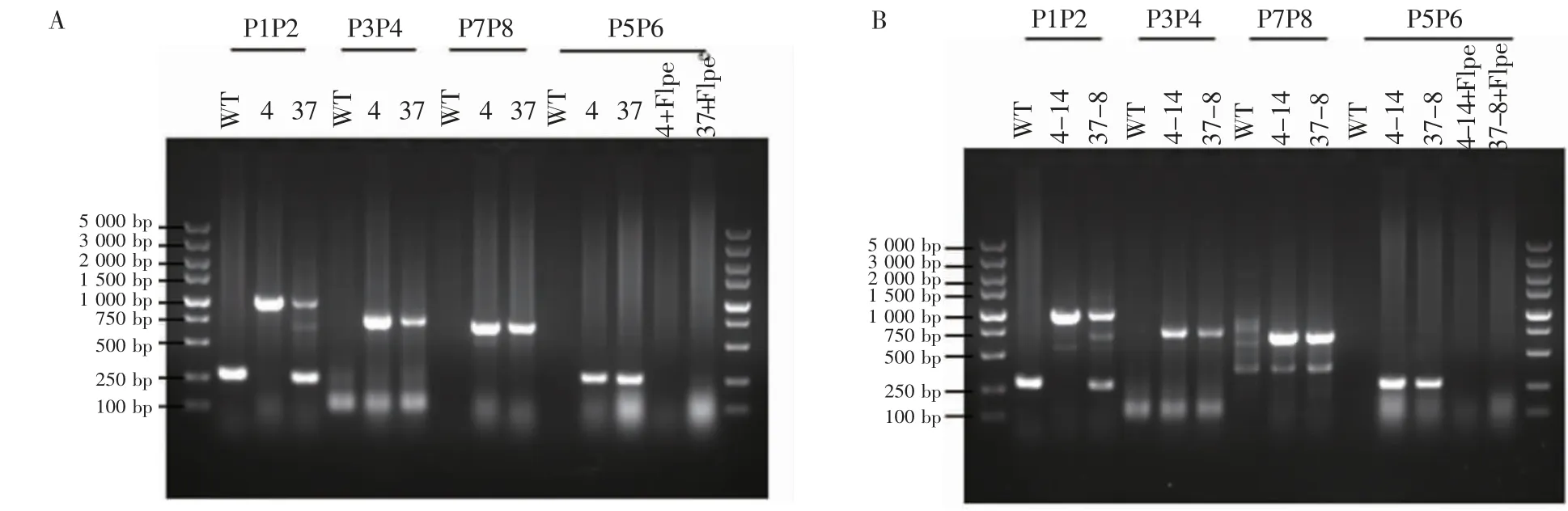
圖4 eGFP-Prdm1/mCherry-Dppa3KnockIn細胞基因型鑒定結果Fig 4 The genotyping results of eGFP-Prdm1/mCherry-Dppa3 Knock In cells
2.4 流式檢測PGCs誘導結果 在進行體外誘導前,即2i+LIF培養條件下,對WT細胞、eGFPPrdm1/mCherry-Dppa3雙雜合子細胞及雙純合子細胞進行流式分析,結果見圖5,WT細胞中未檢測到eGFP和mCherry熒光信號,Knock In細胞中可以檢測到微弱mCherry和eGFP熒光信號,并且純合子的mCherry和eGFP熒光信號比雜合子強,此結果符合在mESCs中Dppa3基因有少量表達,Prdm1基因也有微弱表達。
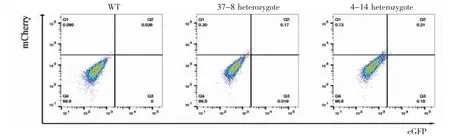
圖5 未誘導時WT及Knock In細胞流式檢測結果Fig 5 Flow cytometry results of WT and Knock In cells without induction
選用WT細胞和eGFP-Prdm1/mCherry-Dppa3 Knock In細胞進行PGCs誘導實驗(實驗流程示意圖見圖6A),使用Activin A、bFGF和1%KSR刺激2 d后,細胞形態從克隆狀變成扁平的上皮樣,說明成功將ESCs誘導為EpiLCs;然后將3×103個細胞轉移到低吸附96孔板中培養,加入500 ng/mL BMP4、500 ng/mL BMP8a、100 ng/mL SCF、50 ng/mL EGF、1 000 U LIF,培養4 d后收集細胞進行流式分析,培養過程見圖6B,流式分析結果見圖7,與WT相比,Knock In細胞出現明顯的eGFP和mCherry陽性,成功由EpiLCs狀態誘導為PGCLCs,成功構建了eGFP-Prdm1/mCherry-Dppa3 Knock In細胞系。
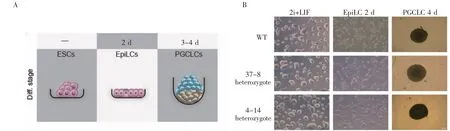
圖6 PGCLCs誘導實驗流程示意圖Fig 6 Schematic diagram of PGCs induction experiment
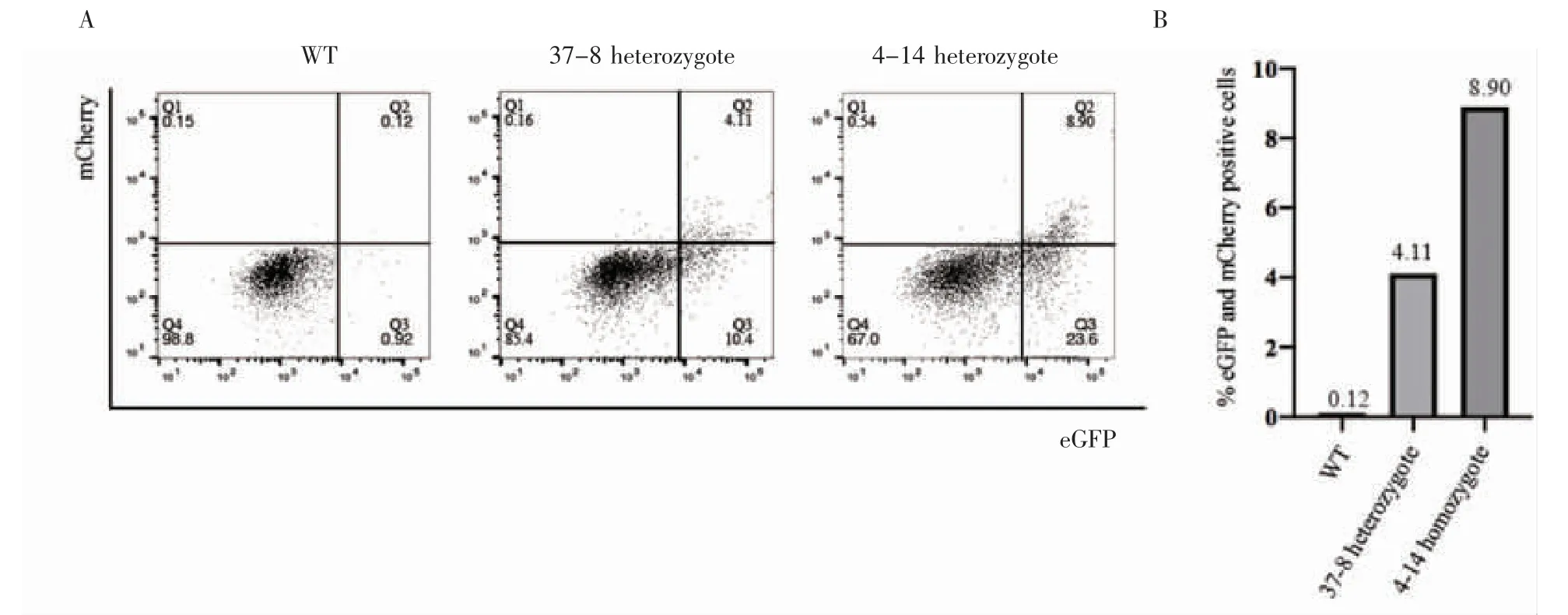
圖7 原始生殖細胞誘導流式檢測結果Fig 7 The flow cytometry results of primordial germ cell induction
3 討論
PGCs是生殖細胞的起源,在早期胚胎發育過程中,PGCs在尿囊底部形成,隨著胚胎發育,大量的PGCs沿著后腸遷移到生殖嵴,最終分化為功能性配子,這種早期譜系選擇依賴于BMP/Smad信號通路激活Prdm1基因表達,從PGCs形成直至遷移到生殖嵴,Prdm1一直持續表達[16]。在小鼠中,Prdm1基因缺失可導致胚胎出現大量的血液滲漏和組織凋亡,并在妊娠中期死亡。Prdm1純合子突變體胚胎無法產生PGCs,雜合子突變體胚胎中PGCs數量顯著減少[17]。Dppa3最初是在小鼠原腸胚時期出現并一直持續表達到E15.5。Dppa3缺陷的雌性個體可以正常受精,但由于染色質壓縮和基因轉錄抑制,很難產生后代[18]。在生殖細胞發育過程中,PGCs在不同的時間點表達不同的基因并進行特定的表觀遺傳重塑。當在小鼠胚胎成纖維細胞中過表達3種特定的生殖細胞基因(Dppa3、Oct4和Nanos2)時,許多與干細胞自我更新以及生殖細胞程序相關的基因會被激活[19],這些都表明Dppa3在生殖細胞發育過程中具有重要作用。
胚胎發育以及相關的遺傳信息通過配子傳遞給后代,因此研究PGCs的特化過程對于理解早期胚胎發育過程以及相關疾病的機制具有重要意義。但是早期胚胎發育過程中,體內PGCs數量十分稀少,這極大影響了對于早期生殖細胞發育進程及相關分子機制的探究,因此需要在體外建立由ESCs分化形成PGCs的培養系統。在之前的研究中,Ohinata等[20]構建Prdm1-Venus及ECFP-Dppa3 BAC(Bacterial artificial chromosome),用顯微注射針將線性化的外源DNA片段注入小鼠受精卵的原核中,從而獲得Prdm1-Venus及ECFP-Dppa3轉基因小鼠,將這兩種小鼠進行后續交配以獲得雙純合小鼠(將雙純合命名為BVSC:Blimp1-Venus and Stella-ECFP),并從與BVSC雄性交配的雌性小鼠囊胚中獲得帶有PGC報告基因的ES細胞系。此方法中外源DNA為隨機整合,不同整合位點的PGC報告基因的表達水平有很大的差異需要通過建系篩選外源基因高表達的系,過程繁瑣,并且外源DNA的隨機整合還可能影響內源基因的表達。此外,真核細胞中的很多順式作用元件與靶基因相距較遠,BAC質粒中可能缺失這些遠程調控元件,因此利用BAC質粒獲得的帶有PGC報告基因的ES細胞系并不一定能真實地反映PGC基因在ES細胞分化過程中的表達。CRISPR-Cas9系統通過非常短小的guideRNA形成靶向特異性,該系統極大地簡化了基因組編輯操作,應用領域涵蓋干細胞工程、基因治療、組織和動物疾病建模等諸多方面。
在本實驗中,利用CRISPR技術實現基因組定點編輯,在mESCs中構建mPGCs分化的報告基因系統,為PGCs提供穩定、可靠和安全的標記方法,有助于后續進一步探究PGCs分化的分子機制,也可促進對早期胚胎發育過程的理解以及不孕不育等相關疾病的研究。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
東方少年·布老虎畫刊(2023年8期)2023-08-01 15:45:12
科學大眾(2021年6期)2021-07-20 07:42:44
科學(2020年3期)2020-11-26 08:18:30
學苑創造·A版(2020年9期)2020-10-13 09:41:02
娃娃樂園·綜合智能(2019年3期)2019-04-03 09:17:36
中成藥(2018年2期)2018-05-09 07:19:34
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
湖北師范大學學報(自然科學版)(2015年2期)2016-01-10 08:41:55
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
