超高效液相色譜-四極桿-飛行時間質譜技術非靶向定性篩查豬肉中79種藥物殘留
2023-01-05 11:16:28王建鳳高麗娟王秋水馮月超
分析測試學報 2022年12期
劉 佳,王建鳳,高麗娟,王秋水,馮月超,劉 艷,丁 奇,王 穎,邵 鵬
(北京市科學技術研究院分析測試研究所(北京市理化分析測試中心),北京 100089)
現代畜牧業(yè)中,獸藥被廣泛用于維持動物健康、預防感染和治療疾病[1]。然而,用藥不規(guī)范或者非法使用違禁藥物將導致動物源性產品中違禁藥物檢出、獸藥殘留超過國家標準規(guī)定,產生食品安全風險[1-4]。為了確保食品安全,國際食品法典委員會、歐盟委員會、中國等均規(guī)定了食品中藥物最大殘留限量[5],如GB 31650-2019[6]《食品安全國家標準-食品中獸藥最大殘留限量》中規(guī)定了動物源性食品中磺胺類抗生素、倍氯米松等的最大殘留限量。考慮到養(yǎng)殖者用藥的不確定性和復雜性,建立一種高通量、快速測定動物源性食品中多藥物殘留的檢測方法刻不容緩。
動物源性食品存在基質復雜、干擾物多等特點,且多藥物分析中藥物極性往往差異較大,單一的前處理技術難以適用于所有目標物。最初用于農藥殘留分析的快速樣品前處理技術——QuEChERS目前已被廣泛用于動物源性食品中多類獸藥的殘留分析[7-9],然而該方法無法充分回收動物養(yǎng)殖中常用的極性藥物,如四環(huán)素和喹諾酮類[5]。因此,建立一種適用于多組分的前處理技術十分必要。
目前,液相色譜-串聯質譜法被廣泛用于動物源性食品中多藥物的同時檢測[7,10-13]。該法具有高的靈敏度和選擇性,但存在兩點不足:一是無法分析多反應監(jiān)測(MRM)中未定義的化合物,因此無法獲得關于未知藥物的信息[14];另一方面,通過串聯質譜進行多藥物殘留分析可能非常耗時,尤其是對數百種不同化合物的分析[15]。四極桿飛行時間質譜(TOF MS)具有數據采集速度快、分辨能力高、質量精度高、檢測靈敏度高等多重優(yōu)點,能夠同時分析無限數量的化合物,實現非靶向目標化合物定性,近年來已發(fā)展成為一種高效的篩查方法,在食品安全分析領域有害物質高通量、快速篩查方面得到廣泛應用[9,14,16-19]。
本研究以GB 31650-2019[6]、《中華人民共和國農業(yè)農村部公告第250號公告》[20]等中限用、禁用的β-興奮劑、蛋白同化激素、抗生素等79種藥物為研究對象,在優(yōu)化樣品前處理技術的基礎上,采用超高效液相色譜-四極桿-飛行時間質譜(UPLC-Q-TOF MS)技術,建立了豬肉中79種藥物的多殘留定性篩查方法。該方法具有通量高、簡便、快速等特點,具有較強的實際應用價值。
1 實驗部分
1.1 儀器、試劑與樣品
Acquity UPLC超高效液相色譜儀、SYNAPT G2-Si四極桿飛行時間質譜儀、Waters Acquity BEH HSS-C18柱(美國Waters公司);GR22GⅢ高速冷凍離心機(日本Hitachi公司);MS200多管渦旋混勻儀(杭州瑞誠儀器有限公司);N-EVAPTM112恒溫水浴氮吹儀(美國Organomation Associates公司);Secura225D-1CN天平(德國Sartorius公司);Vortex Genius 3旋渦混合器(德國IKA公司);Milli-Q超純水儀(美國Millipore公司);FAVEX-NM50獸藥殘留快速柱(500 mg/6 mL,高雄巨研科技股份有限公司)。
79種藥物標準品(純度≥98%)購自德國Dr.Ehrenstorfer公司和美國A Chemtek公司;乙腈、甲醇、甲酸(色譜純)、甲酸銨(美國Thermo Fisher Scientific有限公司);叔丁基甲基醚(色譜純,美國Sigma-Aldrich公司);碳酸鈉(Na2CO3)、乙二胺四乙酸二鈉鹽(EDTA-Na2)(分析純,國藥集團化學試劑有限公司);0.22 μm有機尼龍濾膜(天津津騰公司)。
所用樣品為市售豬肉鮮肉,購自當地超市。
1.2 實驗方法
1.2.1 溶液配制
根據各藥物標準品的溶解性,選擇甲醇、乙腈等溶劑配制質量濃度100~1 000 μg/mL的各藥品單一標準品儲備液,-18℃避光保存。
混合標準儲備液:分別移取適量單一標準品儲備液于10 mL容量瓶中,用甲醇稀釋配制成1 μg/mL的混合標準儲備液。
基質標準工作溶液:取適量混合標準儲備液,用基質空白提取液逐級稀釋,配制成質量濃度分別為0.01、0.05、0.1、0.25、0.5、1、2、5、10、20、50、100、200 μg/L的基質標準工作溶液,現用現配。
1.2.2 樣品制備
1.2.2 .1樣品提取稱取粉碎均質后的樣品2 g(精確至0.01 g)于50 mL聚丙烯離心管中,先加入10 mL 0.5%(體積分數)甲酸-乙腈溶液,充分混勻后以2 500 r/min振蕩提取30 min,10 000 r/min下冷凍離心10 min后將上清液全部移出,再加入10 mL甲醇,以2 500 r/min振蕩提取30 min,10 000 r/min冷凍離心10 min,上清液全部移出。分別準確移取兩次上清液各5 mL,置于FAVEX-NM50凈化柱中,以每秒1滴流速加壓,收集濾液,于40℃氮吹至近干,用1 mL乙腈(0.1%甲酸)∶5 mmol/L甲酸銨(0.1%甲酸)(13∶87,體積比)定容,渦旋復溶,經0.22 μm有機尼龍濾膜過濾,待儀器分析。
1.2.2 .2基質空白提取液的制備取不含被測物的豬肉樣品,按照“1.2.2.1”操作處理,得到基質空白提取液。
1.2.3 分析方法
(1)城鄉(xiāng)發(fā)展聯動性不足。長沙市城鄉(xiāng)旅游產業(yè)結構和要素目前尚不完整,與工業(yè)、農業(yè)、林業(yè)以及其他產業(yè)融合不足:縣域內旅行社、游客集散中心、旅游咨詢中心等功能發(fā)揮的不夠;餐飲住宿、休閑經營規(guī)模有待擴大,服務水平有待提升;缺少規(guī)模化的特色手工藝品、土特產等旅游商品及其加工的銷售場所。同時由于各鎮(zhèn)村各自為政、爭先恐后地做規(guī)劃、出臺優(yōu)惠辦法來發(fā)展鄉(xiāng)村旅游,統籌開發(fā)不足、景點同質化情況日漸增多。
1.2.3 .1色譜條件色譜柱:Waters Acquity BEH HSS-C18(2.1 mm×150 mm,1.7 μm);流動相A:0.1%(體積分數)甲酸-乙腈;流動相B:0.1%(體積分數)甲酸-5 mmol/L甲酸銨溶液。梯度洗脫程序:0~0.5 min,13%A;0.5~10.0 min,13%~50%A;10.0~10.75 min,50%~95%A;10.75~12.25 min,95%A;12.25~12.5 min,95%~13%A;12.5~15.0 min,13%A。柱溫:40℃;進樣量:10 μL;流速:0.4 mL/min。
1.2.3 .2質譜條件離子源:電噴霧離子源(ESI);掃描方式:正離子掃描;離子源溫度:150℃;電噴霧電壓:3 000 V;脫溶劑氣流速:800 L/h;脫溶劑氣溫度:400℃;錐孔氣流速:50 L/h;錐孔電壓:15 V;掃描時間:0~15 min;掃描間隔時間:0.1 s;MSE(全信息串聯質譜)模式檢測,靈敏度模式;質量掃描范圍:m/z50~1 200;數據采集模式:Continuum模式;碰撞能量范圍:10~60 eV。質量校正液為亮氨酸腦啡肽。
1.2.4 數據處理
利用Masslynx軟件對前處理后的樣品進行數據采集,以UNIFI軟件進行數據分析,基于實驗室自行建立的包括79種藥物在內的質譜數據庫[21]進行檢索,以目標化合物的一級質譜精確分子量、保留時間、碎片離子等信息進行藥物殘留的快速定性分析。
2 結果與討論
2.1 色譜條件優(yōu)化
所測定的79種藥物化學性質相差較大,較難達到良好分離,尤其是對于分子式相同或結構式相近的化合物,其有相近或相同的碎片離子,且出峰時間相近易造成譜圖干擾。對于高分辨質譜,目標物質在色譜柱上的分離程度越高,其二級質譜的干擾碎片信息越少,更有利于定性結果的準確判斷[22]。實驗發(fā)現有機相為乙腈時,化合物峰形對稱性好、分離度高、保留較好;當在水相和有機相中加入甲酸時,可明顯提高β-興奮劑、糖皮質激素、蛋白同化激素類化合物在離子源中的離子化效率,增加正離子目標物質的響應值。同時,甲酸銨溶液對部分目標物質的色譜峰形具有改善作用。因此,結合出峰時間、峰形以及化合物響應,選用乙腈(0.1%甲酸)-5 mmol/L甲酸銨(0.1%甲酸)作為流動相,梯度洗脫程序見“1.2.3.1”。
2.2 樣品前處理條件優(yōu)化
進行非靶向篩查時要求前處理方法能夠全面地提取樣品中的化合物[23]。因此,以提取物質的數量盡可能多為前提進行樣品前處理條件優(yōu)化,對提取溶劑進行考察。
2.2.1 提取溶劑優(yōu)化
本研究中化合物的極性范圍非常廣泛,且絕大多數目標化合物在有機溶劑中的溶解度較大,因此分別采用甲醇、乙腈、叔丁基甲基醚中的一種或多種溶劑對目標化合物進行提取。已有研究表明[16,24],提取溶劑的pH值會影響酸堿兩性物質的電離,因此,同時考察了有機溶液中加入弱酸和弱堿對目標物的提取效果。分別研究了①甲醇,②0.5%甲酸-甲醇,③乙腈,④0.5%甲酸-乙腈,⑤甲醇和乙腈等比例混合溶劑(含0.5%甲酸),⑥甲醇、乙腈和0.1%EDTA-Na2等比例混合溶劑(含0.5%甲酸),⑦10%Na2CO3和叔丁基甲基醚(1∶9,體積比)的提取效果。不同提取溶劑下目標化合物的提取情況如表1所示,提取液⑥和⑦分別提取出77種和51種目標化合物,除四環(huán)素類中的土霉素外,提取液③提取出其它78種目標化合物。其余4種提取溶劑均提取出全部79種目標化合物,目標物提取率達100%。在這4種提取溶劑中,79種目標化合物的提取回收率如圖1所示。由于目標化合物性質差別較大,其回收率表現出較大差異:對于大環(huán)內酯類以及磺胺類抗生素,甲醇表現出明顯優(yōu)于其他3種提取溶劑的提取效果;0.5%甲酸-乙腈對大多數喹諾酮類抗生素、β-興奮劑、蛋白同化激素、糖皮質激素以及孕激素拮抗劑的提取效果優(yōu)于其它3種提取溶劑。綜合考慮79種物質的平均回收率和目標化合物在不同回收率范圍的個數(表2),選擇0.5%甲酸-乙腈和甲醇組合作為提取溶劑,并對兩種溶劑的組合順序進行優(yōu)化。
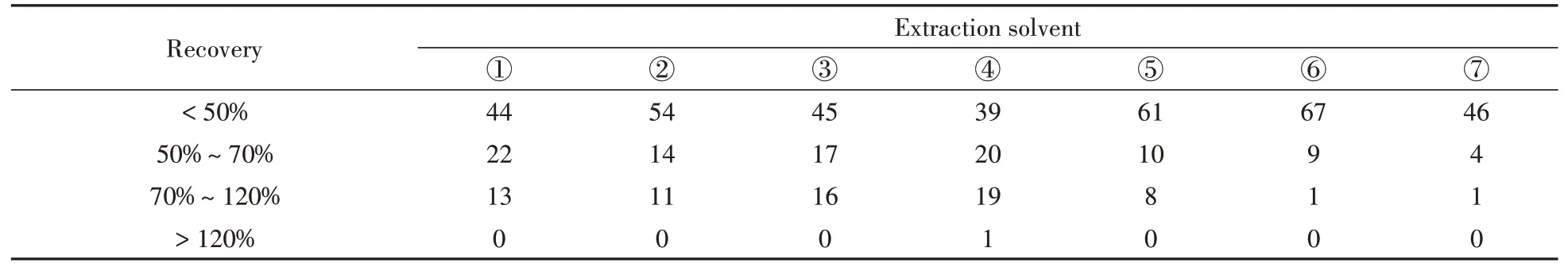
表2 不同提取溶劑下豬肉中79種目標化合物在不同回收率范圍的個數統計Table 2 Statistics of 79 target compounds in pork under different extraction solvents in different recovery ranges

圖1 不同提取溶劑對79種目標化合物回收率的影響Fig.1 Effect of different solvents on the recoveries of 79 target compounds
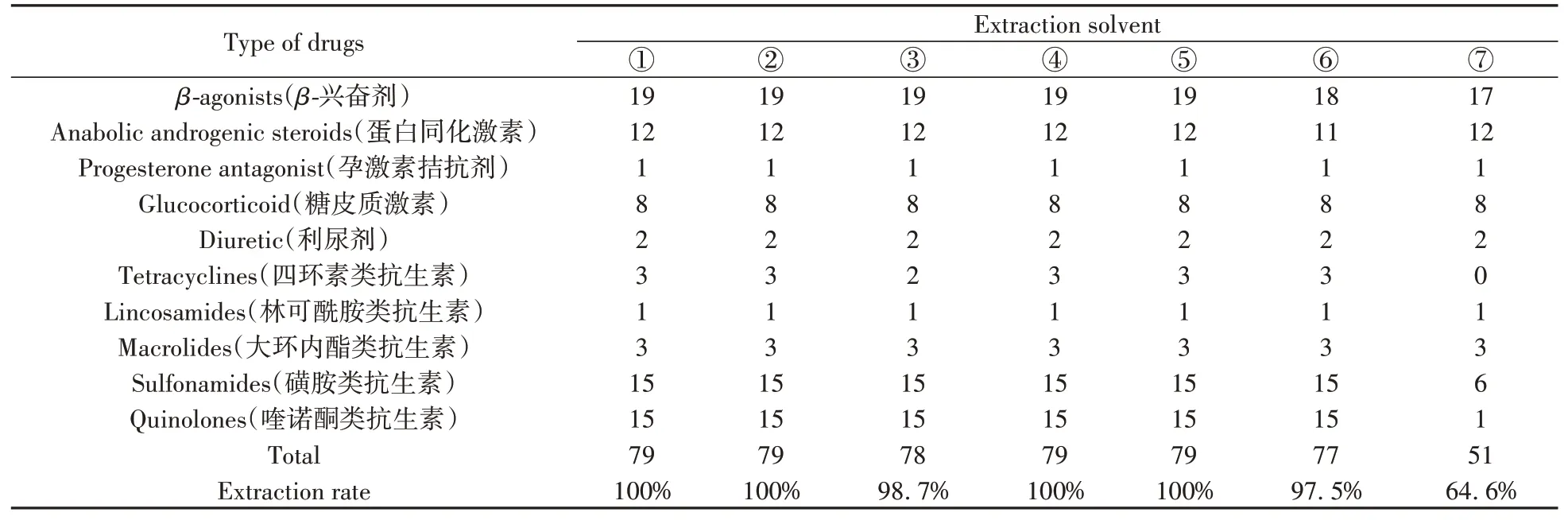
表1 不同提取溶劑對豬肉中79種目標化合物的提取情況Table 1 Extraction of 79 target compounds from pork with different extraction solvents
2.2.2 提取溶劑組合優(yōu)化
由于物質種類較多,每類化合物中選擇2~3種代表性物質,考察①先甲醇后0.5%甲酸-乙腈;②先0.5%甲酸-乙腈后甲醇;③甲醇和0.5%甲酸-乙腈同時提取對回收率的影響,結果如圖2所示。除阿奇霉素、羅紅霉素外,組合②對于選定目標化合物的提取效果優(yōu)于其他兩種組合。因此,采用先0.5%甲酸-乙腈后甲醇的組合提取順序。

圖2 部分目標化合物在不同組合提取順序下的回收率Fig.2 Recoveries of some target compounds under different solvent combinations
2.3 基質效應
基質效應是指目標分析物以外的其他組分引起的分析信號增強或抑制現象[25],通過采用基質標準曲線斜率與溶劑標準曲線斜率的比值(SSE)來評價基質效應[26]。SSE值為0.8~1.2時,表明基質效應不明顯[27];當SSE>1.2時,表明存在基質增強效應;當SSE<0.8時,表明存在基質抑制效應[28]。本實驗分別配制豬肉空白基質標準曲線與對應濃度的溶劑標準曲線考察SSE值,結果見表3。結果表明:86.1%(68/79)的目標物質SEE在0.8~1.2范圍內,6.3%(5/79)的目標物質SEE<0.8,存在基質抑制效應,7.6%(6/79)的目標物質存在基質增強效應。基質效應影響定量結果的準確性,通常采用同位素內標法或基質匹配標準曲線法[22]進行校正。本方法的目標物質接近百種,使用內標的成本偏高,不利于方法的推廣,因此,采用空白基質標準曲線進行校正以降低基質對測定結果的影響。

表3 豬肉中79種藥物的信號抑制/增強、線性方程、線性范圍、相關系數、檢出限和定量下限Table 3 Signal suppression/enhancements(SSEs),regression equations,linear ranges,correlation coefficients(r2),limits of detection(LODs)and limits of quantitation(LOQs)of 79 drugs in pork
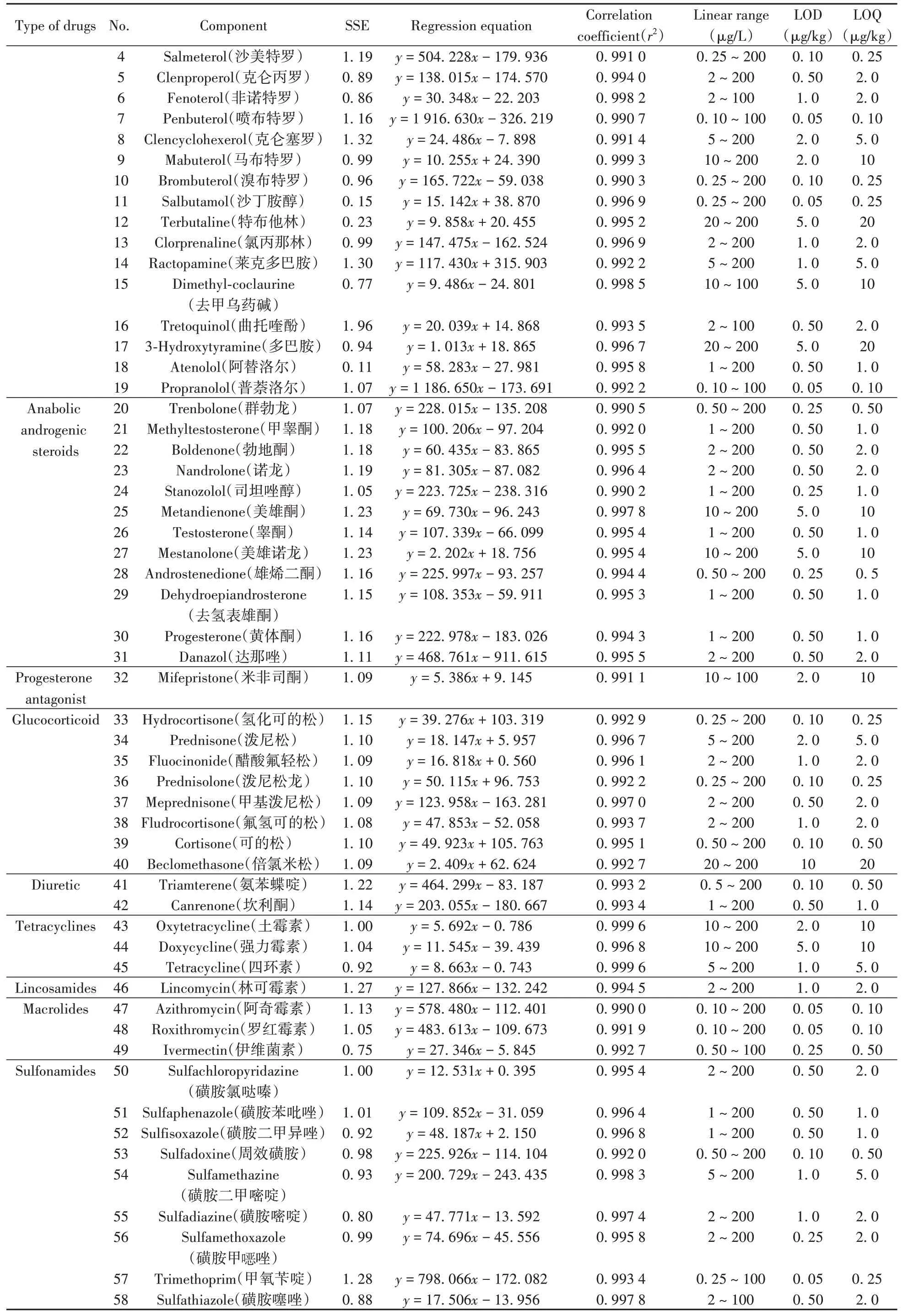
(續(xù)表3)

(續(xù)表3)
2.4 線性關系、檢出限及定量下限
用空白基質提取液配制系列濃度的79種藥物混合標準工作溶液,以峰面積(y)為縱坐標,對應的質量濃度(x,μg/L)為橫坐標,繪制基質標準工作曲線,79種藥物在相應質量濃度范圍內線性關系良好,相關系數(r2)均不低于0.99(表3)。分別以3倍信噪比(S/N)和10倍信噪比對應的濃度確定方法的檢出限(LOD)和定量下限(LOQ)[28],79種藥物的LOD為0.05~10 μg/kg,LOQ為0.10~20 μg/kg。通過與GB 31650-2019[6]中提及的相關藥物殘留限量相比,該方法定量下限遠低于對應物質限量標準,可以很好地滿足食品中藥物殘留的篩查需求。
2.5 非靶向篩查技術的應用
2.5.1 模擬加標樣本分析
選取每類物質中的1~3種代表性物質共21種(普萘洛爾、群勃龍、勃地酮、諾龍、米非司酮、雄烯二酮、氫化可的松、潑尼松、去氫表雄酮、可的松、倍氯米松、氨苯蝶啶、阿奇霉素、羅紅霉素、伊維菌素、磺胺二甲嘧啶、磺胺嘧啶、甲氧芐啶、磺胺甲噻二唑、氧氟沙星、奧比沙星),采用所建立的方法對加標水平為10 μg/kg和20 μg/kg的上述21種物質的豬肉樣本進行檢測。通過檢索質譜信息數據庫,采用精確質量數和保留時間的方式進行非靶向定性篩查,在質量數精確度允許偏差為10 ppm,保留時間偏差為0.2 min,且至少檢測到1種碎片離子的條件下,10 μg/kg加標水平的篩查結果見表4,部分典型藥物的全掃描圖見圖3。由表4可知,10 μg/kg加標下,除定量下限高于10 μg/kg的倍氯米松(表3)未篩查出外,其余物質全部篩出;20 μg/kg加標下21種物質可全部篩查出來。

圖3 部分典型藥物的全掃描圖Fig.3 Full-scan chromatograms of some typical drugs

表4 10 μg/kg加標水平下樣本的篩查結果Table 4 Screening results of specimen at 10 μg/kg spiked level

(續(xù)表4)
2.5.2 實際樣本檢測
采用本方法對市售的6份豬肉樣本進行篩查分析,結果顯示,除1份樣本為陰性外,其余5份樣本均檢出氫化可的松,含量為1.39~13.6 μg/kg。所有陽性樣本中氫化可的松的離子加和模式均為[M+H]+。為了檢驗篩查結果的準確性,采用高靈敏度Xevo TQ-S串聯四極桿質譜[8]對陽性樣本進行驗證,以樣品3為例,Xevo TQ-S串聯四極桿質譜儀的確證色譜圖見圖4。與本方法結果一致。
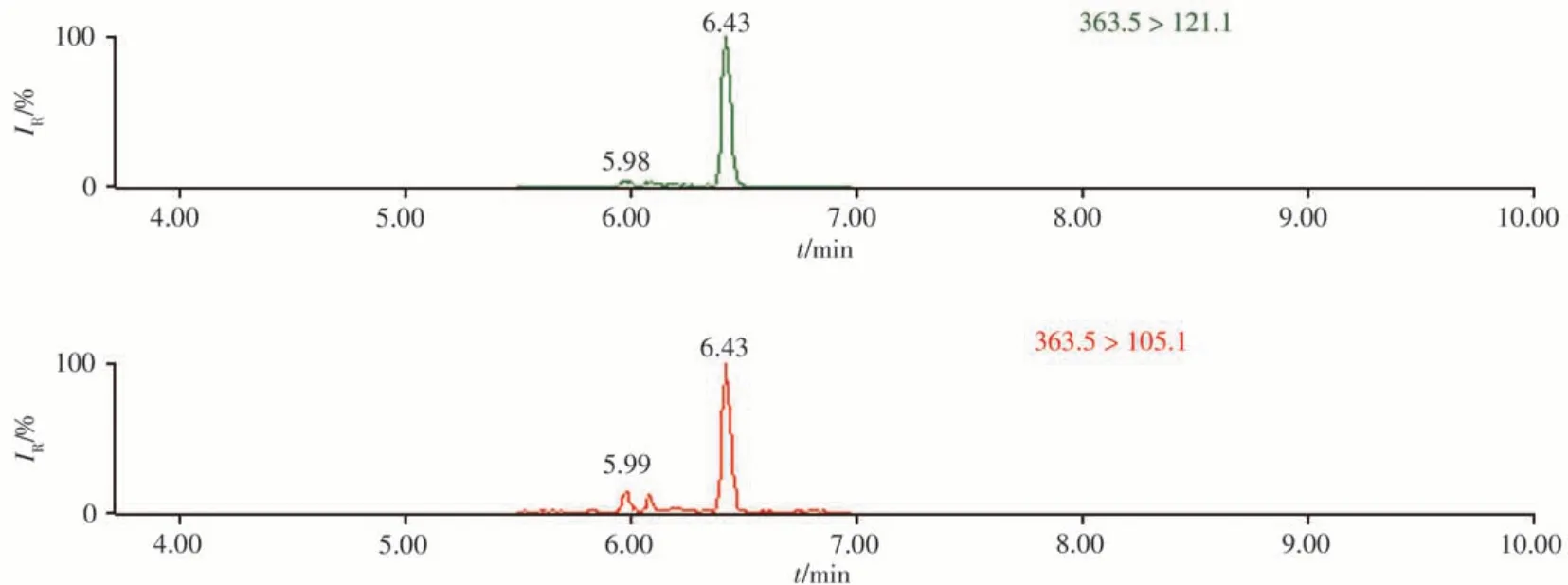
圖4 樣品3的Xevo TQ-S串聯四極桿質譜儀色譜圖Fig.4 Xevo TQ-S tandem quadrupole mass spectrometer chromatogram of sample 3
我國GB 31650-2019[6]中規(guī)定,氫化可的松是被允許外用于所有食品動物且不需要制定殘留限量的獸藥。結果表明6份市售豬肉樣本在所研究的79種藥物范圍內是安全的。
3 結論
本研究基于UPLC-Q-TOF MS,建立了無需標準品即可實現豬肉中79種藥物殘留的快速定性篩查技術,并將其應用于模擬陽性樣本以及隨機市售樣本的篩查。結果表明該方法具有通量高、簡便、快速、高效等優(yōu)點,可用于風險預警、日常監(jiān)測及應急檢測,為豬肉類食品安全提供了有力的技術支撐。
