茶網蝽線粒體基因組全序列測定及系統發育分析
2023-01-11 07:43:02楊金宏謝滿超文欣茹陳蕊茹孔衛青
茶葉科學 2022年6期
關鍵詞:結構
楊金宏,謝滿超,文欣茹,陳蕊茹,孔衛青
1. 安康學院現代農業與生物科技學院/陜西省茶葉省市共建重點實驗室,陜西 安康 725000;2. 安康學院陜西省蠶桑重點實驗室,陜西 安康 725000
茶網蝽(Stephanitis chinensis)為半翅目(Hemiptera)異翅亞目(Heteroptera)網蝽科(Tingidae)昆蟲,不完全變態,以我國西南地區的油茶和茶樹等經濟作物為主要寄主。茶網蝽若蟲和成蟲以刺吸式口器從茶樹樹冠中、下層成熟葉片背面吸食汁液,其排泄物在葉片上形成黑色黏質物[1],受害茶樹樹勢衰弱,生長緩慢,發芽率低且芽包小,嚴重影響茶樹生長和茶葉產量[2]。
茶網蝽在陜南地區的茶葉產區一年發生2~3代,繁殖力強,群體數量龐大,種群擴散能力強,新舊茶園均受危害。2010年陜西省漢中市首次報道茶園發生茶網蝽危害[3],2012年擴散至陜西省安康市,2017年蔓延至漢江流域和嘉陵江流域[4]。茶網蝽成蟲體長 3.1~4.2 mm,體小扁平,無趨性,初孵若蟲肉眼無法識別,亟待分子生物學鑒定方法[5]。
線粒體基因組(Mitochondrial genome,mitogenome)不易發生重組、倒位、易位等突變,結構保守。線粒體基因進化速率快,為核基因的 2~6倍,被廣泛應用于種群多樣性鑒定、物種系統發育、物種地理學譜系和系統發生學等方面的研究[6-7]。全世界已知網蝽科昆蟲有260屬2 124種,但目前NCBI公布的線粒體基因組僅27個(截至2022年3月31號),網蝽科昆蟲的線粒體基因組大小差異較大,主要是由于控制區(Control region)數量、長度不等的重復序列造成[8]。本研究利用Illumina和Sanger測序,獲得了陜西安康茶區茶網蝽的線粒體基因組全序列及特征,并進行了網蝽科的系統發育分析,旨在為進一步對茶網蝽擴散傳播途徑、防治及異翅亞目的分類研究提供依據。
1 材料與方法
1.1 試驗材料
茶網蝽成蟲于2021年5月17日采集自陜西省安康市紫陽縣和平茶廠茶園,采集的樣本存放在無水乙醇中,置于-20℃冰箱保存備用。
1.2 基因組DNA提取與高通量測序
取乙醇浸泡的茶網蝽單個個體,ddH2O沖洗2遍,然后使用快速DNA提取檢測試劑盒[KG203,天根生化科技(北京)有限公司]按照說明書提取基因組 DNA,最后用100 μL ddH2O洗脫。使用 Multiskan FC微量酶標儀(美國Thermo Fisher Scientific公司)對提取的基因組DNA進行檢測。選取部分檢測合格的基因組DNA委托生工生物工程(上海)股份有限公司進行建庫和Illumina Hiseq 2500平臺雙向高通量測序,測序長度為150 bp,目標數據量4 Gb。
1.3 序列拼接組裝與控制區的PCR擴增
對Illumina高通量測序數據進行質控,去除接頭和帶 N的序列,滑窗法去除得分低于20和長度小于 35 bp的序列。最后數據利用GetOrganelle[9](K=31、55、85)和NOVOPlasty[10](K=31)軟件進行組裝,組裝結果使用Bandage[11]進行可視化分析,手動去除可信度低和不確定的部分。在獲得的序列缺口處設計引物rrnS-F 和 trnM-R(表1),使用 Gloria Nova HS 2×高保真Taq DNA聚合酶(RK20715,武漢愛博泰克生物科技有限公司)進行 PCR擴增,PCR反應程序:94℃預變性 2 min;94℃變性 30 s,55℃退火 30 s,72℃延伸 5 min,30個循環;72℃延伸10 min。使用1%瓊脂糖凝膠電泳檢測PCR產物,之后對產物進行 Sanger雙向測序,并根據獲得序列設計測序引物(表1)進行Gap補齊。

表1 引物序列Table 1 Primers sequence
1.4 基因組注釋和分析
使用在線軟件MITOS2[12](http://mitos2.bioinf.uni-leipzig.de/index.py)對茶網蝽線粒體基因組進行注釋,遺傳密碼選擇5(Invertebrate)。同時利用blastn和blastp比對網蝽科其他昆蟲的線粒體基因組數據確定蛋白質編碼基因(Protein-coding genes,PCGs)、tRNA 和 rRNA的邊界位置。最后的注釋文件提交至NCBI數據庫,并利用在線 OGDRAW 軟件[13](https://chlorobox.mpimp-golm.mpg.de/OGDr aw.html)繪制結構圖。tRNA的二級結構預測使用在線軟件tRNAscan-SE[14](http://lowelab.ucsc.edu/tRNAscan-SE),未成功預測的二級結構進行手工繪制。
使用在線工具 Tandem Repeats Finder[15](https://tandem.bu.edu/trf/trf.html)查找控制區中的串聯重復序列,使用MISA[16]檢測其中的簡單重復序列(Simple sequence repeats,SSRs),參數設置為1~6堿基單元的最少重復數分別為10、5、4、3、3、3個。莖環結構預測使用在線工具MFold[17](http://www.unafold.org/mfold/applications/rna-folding-form.php)。
使用 Mega 11軟件[18]統計基因組及各區域 4種堿基的組成和蛋白質編碼基因的密碼子情況,并通過公式AT skew=(A-T)/(A+T)和 GC skew=(G-C)/(G+C)[19]計算核苷酸組成的偏向性。
1.5 系統發育分析
從 NCBI數據庫下載異翅亞目昆蟲線粒體基因組,分別提取13個PCGs序列,然后利用 macse_v2.05軟件[20]基于編碼框架模式對序列進行比對,并把對齊的DNA合并成一個序列。利用 raxmlHPC[21]構建極大似然(Maximum likelihood,ML)發育樹,GTRGAMMA模式,抽樣 1 000次計算bootstrap值,展示高于50%的位點。
2 結果與分析
2.1 茶網蝽線粒體基因組的測序與組裝
Illumina測序數據經質控,獲得2 025 131條序列,GetOrganelle和NOVOPlasty組裝軟件獲得的線粒體基因組序列完全一致的部分長度為14 561 bp,平均深度為415.04倍。控制區序列的組裝得到多種結果,可能存在重復結構。進一步利用引物rrnS-F和trnM-R進行PCR擴增,獲得1條4 000 bp左右的條帶(圖1)。使用設計的多條測序引物對控制區進行Sanger測序,拼接測序結果,獲得序列長度為4 040 bp,與Illumina測序組裝數據拼接成環,獲得茶網蝽線粒體基因組全長為 18 085 bp(GenBank登錄號:OM397399)。

圖1 rrnS-F和trnM-R引物的PCR擴增Fig. 1 The PCR amplification of rrnS-F and trnM-R
2.2 茶網蝽線粒體基因組注釋與結構
分析發現,茶網蝽線粒體基因組為典型的雙鏈閉合環狀結構,包含37個基因,分別為13個PCGs、22個tRNA基因和2個rRNA基因(圖2,表2)。基因組控制區為3 678 bp,其后為trnI(GAU)、trnQ(UUG)、trnM(CAU)和nad2基因,與昆蟲線粒體祖先基因順序(Ancestral gene order)相同[22]。
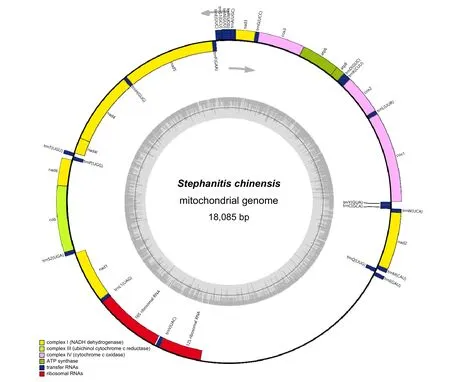
圖2 茶網蝽線粒體基因組圖譜Fig. 2 Map of S. chinensis mitogenome
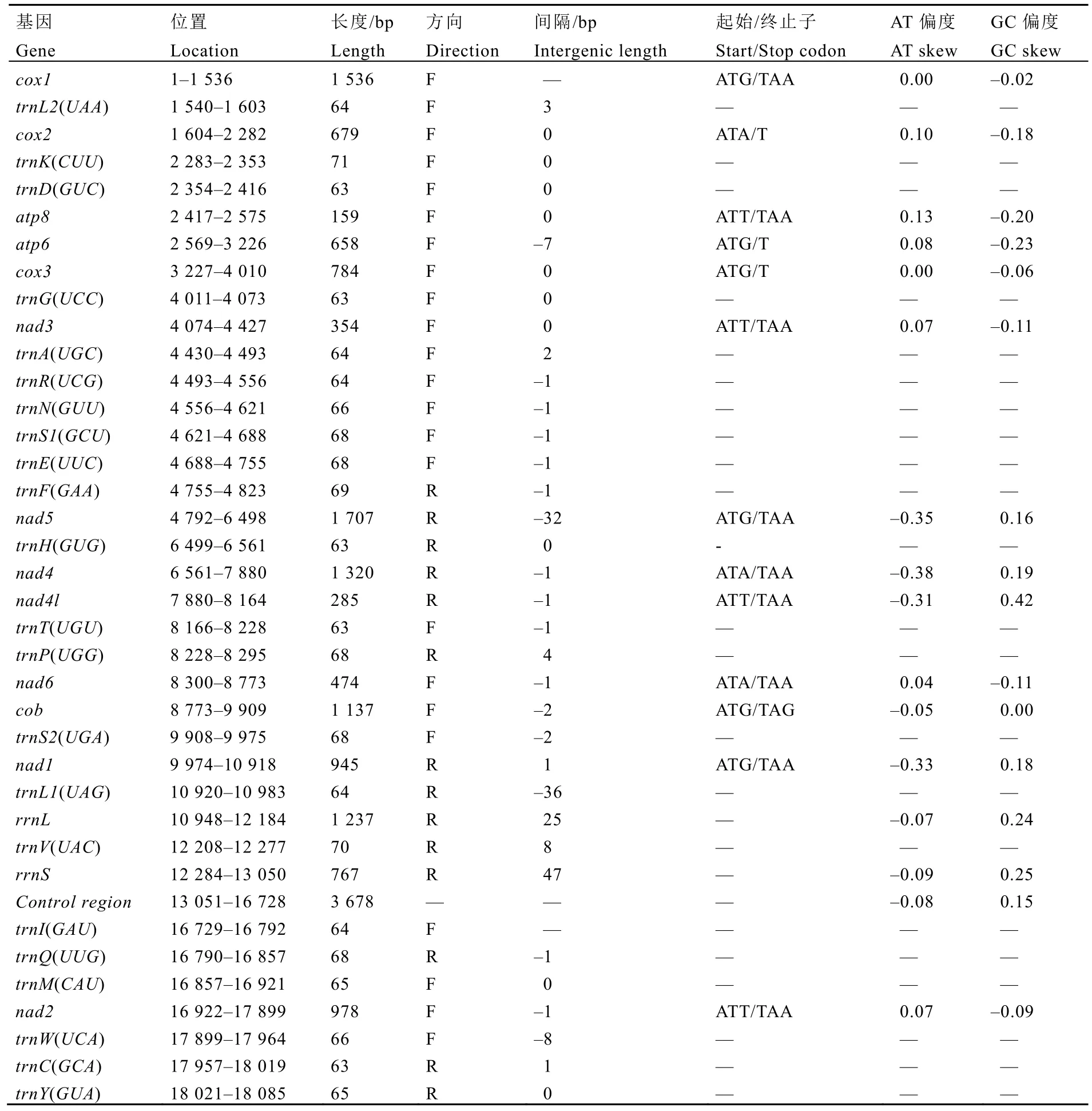
表2 茶網蝽線粒體基因組結構Table 2 The mitogenome organization of S. chinensis
茶網蝽的線粒體基因組的37個基因共存在18處基因重疊,最長的兩處位于nad1和trnL1(UAG)、trnF(GAA)和nad5之間,重疊長度分別為-36 bp和-32 bp,其他位置的重疊長度均在-8 bp以內。基因間隔現象少于基因重疊,37個基因僅存在8處間隔,其中最長兩處位于trnV(UAC)和rrnS、trnL1(UAG)和rrnL之間,長度分別為47 bp和25 bp,其他間隔最長8 bp,另有無間隔和重疊的區域10處(表2)。
2.3 茶網蝽線粒體基因組核苷酸組成
茶網蝽線粒體基因組的 AT含量為78.09%,具有明顯的AT偏好性(表3)。rRNA基因的AT含量最高,為80.84%,AT富集區為79.99%。PCGs的AT含量最低,為76.76%,其中atp8基因的AT含量最高,達84.28%,其次是nad2和nad4l,分別為 82.21%和80.70%;AT含量最低的是cox1和cob,分別為69.08%和72.21%。核苷酸組成在線粒體基因組不同鏈間也不對稱,多數編碼基因所在的J鏈(Majority strand)的PCGs多為AT偏移和CG偏移,而N鏈(Minority strand)的PCGs和rRNA基因,為TA偏移和GC偏移(表2)。
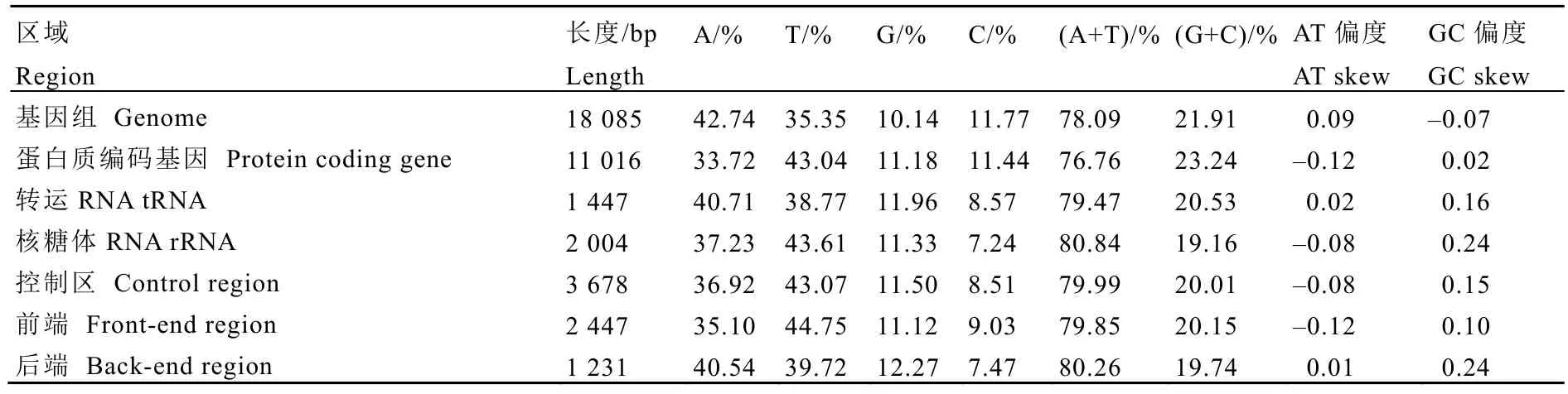
表3 茶網蝽線粒體基因組核苷酸組成Table 3 Nucleotide composition of the mitogenome of S. chinensis
2.4 蛋白質編碼基因
茶網蝽線粒體基因組的13個PCGs的長度在 159~1 536 bp,其中nad1、nad4、nad4l和nad5位于基因組N鏈,其他9個均位于J鏈(表2)。13個基因中,cox1、cox3、atp6、nad5、cob和nad1以ATG起始,atp8、nad3、nad4l和nad2以 ATT起始,cox2、nad4和nad6以ATA起始;以TAA為終止密碼子的基因最多,共有9個,cox2、atp6和cox3以不完全的T終止,cob以TAG終止。同時,PCGs之間存在相互重疊的現象,atp8和atp6基因間重疊了7 bp,nad4和nad4l基因、nad6和cob基因分別重疊了1 bp和2 bp。
進一步分析發現,茶網蝽線粒體PCGs使用最多的 5個密碼子分別是 UUA、AUU、UUU、AUA和 AAU,均由 A和 T組成(表4)。編碼蛋白的氨基酸組成及含量也有較大差異,使用頻率最高的為亮氨酸(Leu),占比約14.00%,其次為異亮氨酸(Ile)、苯丙氨酸(Phe)、絲氨酸(Ser)、甲硫氨酸(Met)、天冬酰胺(Asn)。其他氨基酸占比均低于5%,其中精氨酸(Arg)、半胱氨酸(Cys)、谷氨酰胺(Gln)、組氨酸(His)、天冬氨酸(Asp)的占比低于2%。
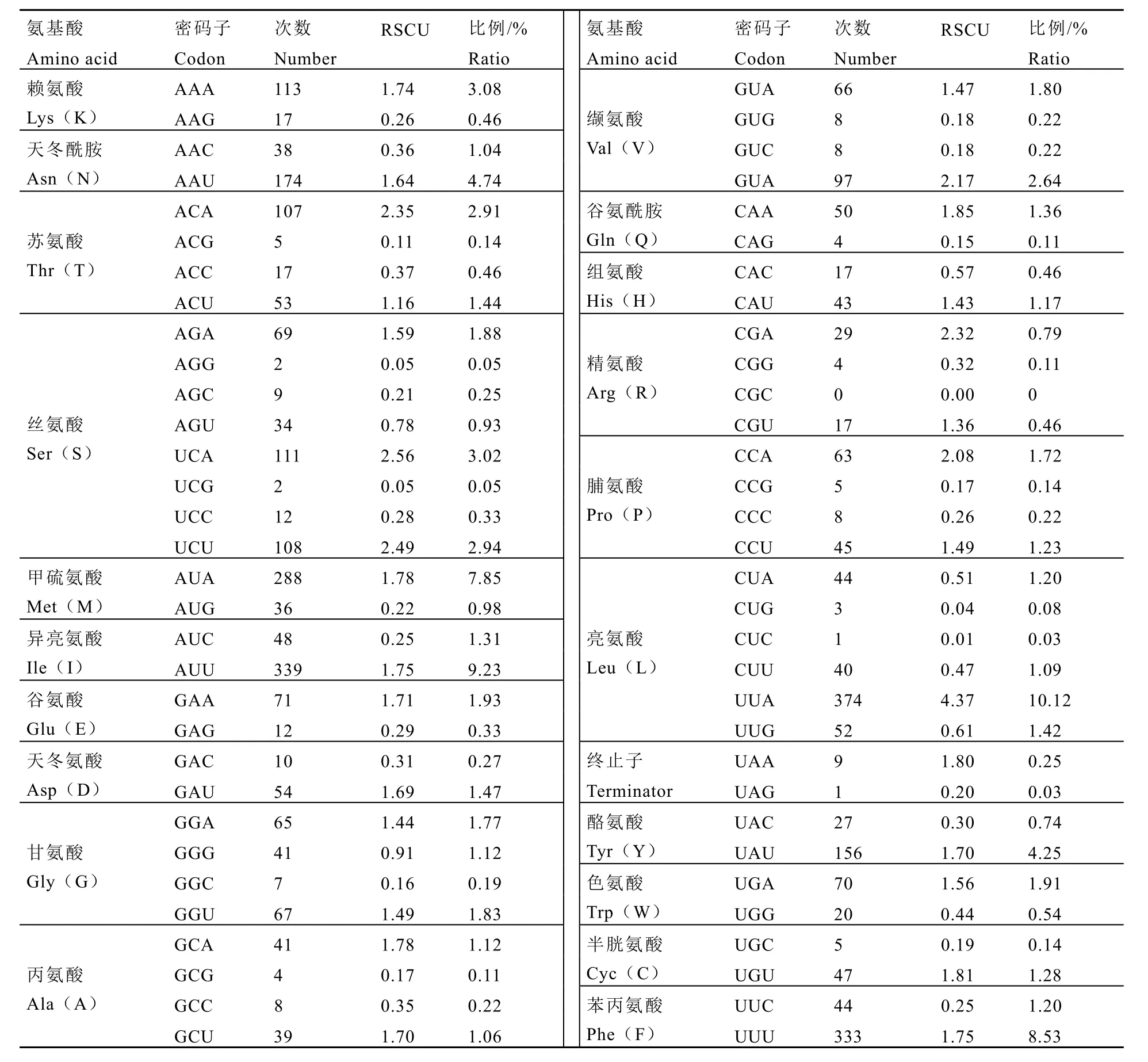
表4 茶網蝽線粒體基因組蛋白編碼基因相對同義密碼子使用度Table 4 Relative synonymous codon usage (RSCU) in the mitogenome of S. chinensis
2.5 tRNA和rRNA基因
茶網蝽線粒體基因組包含22個tRNA基因(圖3),長度在63~70 bp。8個tRNA基因(trnC,trnF,trnH,trnL1,trnP,trnQ,trnV,trnY)位于基因組N鏈,其余14個位于J鏈(表2)。預測 tRNA基因的二級結構發現,trnS1(GCU)缺少DHU臂,其他tRNA均能形成典型的三葉草結構(圖3),該現象存在于多數后生動物中[23-24]。同時,部分tRNA的二級結構還存在G-U和U-U等非經典配對,該現象在其他昆蟲類群 tRNA二級結構中也較為常見[25-26]。
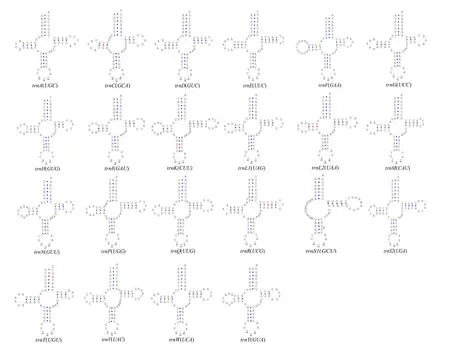
圖3 茶網蝽線粒體基因組tRNA預測二級結構Fig. 3 Predicted secondary structures of tRNA genes in mitogenome of S. chinensis
茶網蝽線粒體基因組含有 2個 rRNA基因,rrnL位于trnL1(UAG)和trnV(UAC)之間,rrnS位于trnV(UAC)和控制區之間,長度分別為1 237 bp和767 bp,AT含量分別為80.22%和81.88%,具有明顯的AT偏向性。
2.6 控制區分析
茶網蝽線粒體基因組的控制區位于rrnS和trnI(GAU)基因之間,長度為 3 678 bp,根據結構將其分為兩部分(圖4)。第一部分位于序列的前端(1~2 447 bp,front-end region,FER),存在以下結構:(1)3組非串聯重復序列。第一組長度為1 540 bp(R1),包含2個498 bp的完整重復單元和 2個該單元的部分序列(477 bp和 63 bp);第二組長度為 154 bp(R2),包含2個77 bp的完整重復單元;第三組長度為582 bp(R3),由3個194 bp的完整重復單元組成。(2)4處(TTAG)n,其中(TTAG)7和(TTAG)9分別連接非串聯重復 R2的 2個單元,另 2處均為(TTAG)5,位于 R2重復的2個單元中。第二部分位于序列的后端(2 448~3 678 bp,back-end region,BER),為串聯重復區(R4),包含5個長度為237 bp的完整重復單元和1個該單元的部分序列。同時序列存在5種莖環結構,其中2種5處為單一環結構,另3種11處為多環結構。
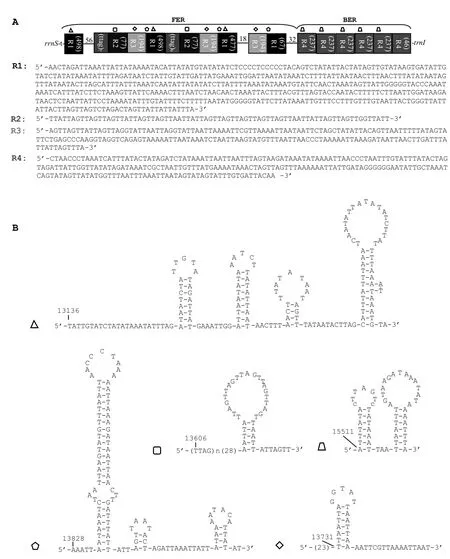
圖4 茶網蝽線粒體基因組控制區結構Fig. 4 Structure of mitogenome control region of S. chinensis
進一步分析該區域的AT含量為79.99%,高于Pseudacysta perseae、Agramma hupehanum和楊柳網蝽(Metasalis populi)該區的AT含量,低于其他網蝽科昆蟲。FER和BER的AT含量分別為79.85%和80.26%(表3),兩段區域對不同核苷酸的使用偏好也有所不同,FER的 AT偏度為-0.12,發生 TA偏移,BER的AT使用量相當,有GC偏移傾向。
2.7 系統發育分析
以雙線銅翅沫蟬(Aeneolamia contigua)為外群,構建了基于異翅亞目41種昆蟲的線粒體PCGs的系統發育樹(圖5)。結果表明,茶網蝽與直脊冠網蝽(Stephanitis mendica)的親緣關系最近,其次為楊柳網蝽和Pseudacysta perseae。網蝽科聚為一簇,位于發育樹的根部,是臭蟲次目(Cimicomorpha)盲蝽科(Miridae)、花蝽科(Anthocoridae)、姬蝽科(Nabidae),以及蝽次目(Pentatomomorpha)、黽蝽次目(Gerromorpha)、歇蝽次目(Nepomorpha)、奇蝽次目(Enicocephalomorpha)的姐妹群。奇蝽次目和黽蝽次目距離最近,與歇蝽次目共同聚合在一簇,蝽次目單獨聚合為一個分支。系統發育樹支持了歇蝽次目、奇蝽次目、黽蝽次目和蝽次目的單系性,而臭蟲次目為并系群。
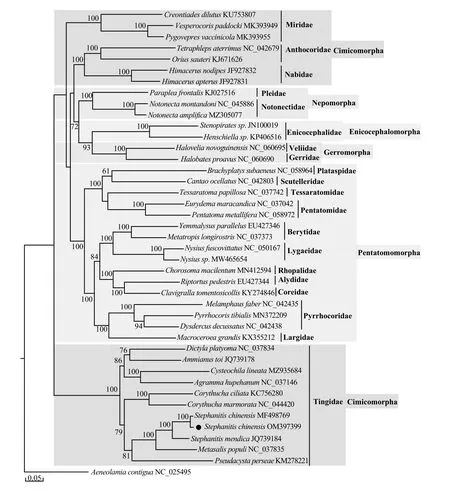
圖5 基于41種昆蟲線粒體蛋白質編碼基因序列構建的ML系統發育樹Fig. 5 Maximum likelihood phylogenetic tree of 41 insects based on the PCG sequences of mitogenome
3 討論
本研究結合Illumina測序和Sanger測序,獲得安康市茶網蝽線粒體基因組全長18 085 bp,是目前 NCBI登錄的網蝽科昆蟲線粒體全序列(15 208~16 667 bp)中長度最長的。控制區是昆蟲線粒體基因組大小變異的主要來源,一般與線粒體基因組的大小呈正相關,控制區的長度差異可以發生在不同物種甚至不同個體之間[27]。目前昆蟲中已報道的最短的控制區僅為70 bp,而象鼻蟲的控制區長度達 9~13 kb[28]。已報道的網蝽科昆蟲線粒體基因組的控制區為733~2 215 bp,而本研究中安康市茶網蝽的控制區為3 678 bp。
控制區是線粒體基因組復制和轉錄的主要調控區,包含線粒體基因組復制相關信息[29]。黽蝽科圓臀大黽蝽(Aquarius paludum)線粒體基因組控制區存在 TATA motif、poly T stretch、莖環結構等保守結構元件[30],地長蝽科大黑毛肩長蝽(Neolethaeus assamensis)的線粒體基因組控制區存在poly A結構、莖環結構、串聯重復區等[31]。本研究發現,茶網蝽基因組中有多種重復序列和莖環結構,而重慶市茶網蝽[32]僅有 R4重復序列,沒有 FER區,對于不同地域茶網蝽基因組之間的差異,有待進一步研究。
本研究茶網蝽的線粒體基因組所有區域均有明顯的 AT偏好性,AT含量最高的是rRNA,這與網蝽科昆蟲楊柳網蝽、菊方翅網蝽(Corythucha marmorata)、古無孔網蝽(Dictyla platyoma)中的情況相同。蛋白質編碼基因的AT含量最低,這有利于物種編碼其所需蛋白質的功能[33]。cox1基因是常用的DNA條形碼基因,應用于昆蟲的識別鑒定[34]。相對較高的 GC含量有利于降低基因的突變率,保持基因穩定性。Yang等[8]研究發現,網蝽科昆蟲線粒體cox1基因的進化速率較低,atp8基因最高,本研究中安康市茶網蝽cox1基因的GC含量最高,atp8基因的GC含量最低,與其結論一致。與多數動物一樣,茶網蝽線粒體基因組不同鏈的核苷酸組成也不對稱[19]。
網蝽科昆蟲線粒體基因組中絕大部分蛋白基因以ATN為起始密碼子,使用最多的是ATG,其中cox1和atp6基因的起始密碼子均為 ATG,ATA的使用頻次略低于 ATT。終止密碼子以TAA為主,cox1、atp8、nad6和nad1基因的終止密碼子都是TAA;其次為T,cox2基因均以單個T作為終止密碼子,cox3和nad5基因使用 T作為終止密碼子的現象較多,較少使用 TA或 TAG。昆蟲線粒體基因組以 T或TA作為終止密碼子現象比較常見,推測其通過轉錄后 mRNA多腺苷酸化形成完全終止密碼子TAA[35]。
臭蟲次目是異翅亞目最大的次目,有 17個科超過20 000種昆蟲,其系統發育研究目前沒有一致的結論。利用形態性狀、轉錄組和核基因等研究發現,臭蟲次目昆蟲為單系[36-38],而使用臭蟲次目昆蟲線粒體基因組數據進行系統發育研究,鑒定其為并系群,尤其是網蝽科昆蟲的數據,該現象更顯著[8,39-40]。Liu等[39]基于線粒體基因組數據分析異翅亞目的系統發育發現,網蝽科和奇蝽次目、鞭蝽次目、黽蝽次目聚為一簇且接近發育樹的根部,是其他異翅亞目昆蟲的姐妹群。本研究利用線粒體PCGs進行的系統發育重建結果,能很好地支持奇蝽次目、黽蝽次目、歇蝽次目和蝽次目的單系性。但臭蟲次目的情況比較復雜,其姬蝽科和花蝽科聚合在一起,形成與奇蝽次目、黽蝽次目和歇蝽次目并列的分支,而其盲蝽科和網蝽科的位置接近發育樹的根部,這與Liu等[39]的研究結果相似。但本研究接近發育樹根部的幾個分支的支持率均低于50%,這可能與網蝽科昆蟲線粒體基因組序列的變異率較大有關[8,39],較大堿基差異不容易區分系統發育關系,更適合于親緣關系近的物種的分化檢測。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
