生酮飲食治療Rett綜合征1例
2023-01-15 04:17:50劉維亮貴州醫(yī)科大學(xué)附屬醫(yī)院兒科貴陽550004貴州醫(yī)科大學(xué)附屬醫(yī)院眼科通訊作者mailliuweiliang05aliyuncom
山西醫(yī)科大學(xué)學(xué)報 2022年12期
關(guān)鍵詞:癲癇
劉維亮,李 芳,艾 戎(貴州醫(yī)科大學(xué)附屬醫(yī)院兒科,貴陽 550004;貴州醫(yī)科大學(xué)附屬醫(yī)院眼科;通訊作者,E-mail:liuweiliang05@aliyun.com)
Rett綜合征(Rett syndrome,RTT)(在線《人類孟德爾遺傳》編號:#312750)是一種神經(jīng)發(fā)育障礙性疾病。RTT呈X連鎖顯性遺傳,本病主要累及女性,男性患者罕見,RTT在女性中的發(fā)病率約為1 ∶15 000~1 ∶10 000,X染色體上的甲基化CpG結(jié)合蛋白2基因(methyl-CpG binding protein 2,MeCP2)是其主要的致病基因[1]。典型RTT患者在6~18個月之前智力運(yùn)動基本發(fā)育正常,之后發(fā)育停止,接著是智力運(yùn)動退化,然后逐漸喪失語言和溝通技能,并喪失獲得的運(yùn)動技能且經(jīng)常出現(xiàn)手部刻板的動作,可出現(xiàn)小頭畸形、癲癇發(fā)作、自閉癥、共濟(jì)失調(diào)、間歇性過度通氣和脊柱側(cè)凸等[2-5]。非典型RTT患者僅具有部分典型RTT患者臨床特征并且缺乏經(jīng)典RTT的進(jìn)展階段[6]。RTT臨床特征包括智力低下、語言功能喪失、手部刻板動作、步態(tài)異常等,該病治療困難,為世界性難題,目前世界范圍內(nèi)僅僅對癥治療,目前臨床尚無治愈手段,以改善癥狀為主。我們使用生酮飲食(ketogenic diet, KD)治療RTT,患者在運(yùn)動、智力方面有一定改善,為國內(nèi)首次系統(tǒng)詳細(xì)報道,特總結(jié)如下。
1 病例報告
1.1 研究對象
患者,女,3歲8月,因“發(fā)育落后3年,抽搐13 d后發(fā)育倒退4 d”入院。患者2月笑,3月抬頭,從不會認(rèn)人,6月時四肢軟,9月坐,4月會抓,1歲手不能抓物體,有反復(fù)洗手動作,吐口水,右腿涼,不會爬,1歲時穿矯形鞋可站立,1歲5月至今不能獨(dú)坐、站立,1.5歲至今僅發(fā)“ba”音,期間外院多家醫(yī)院按“發(fā)育落后”正規(guī)康復(fù)治療至今無好轉(zhuǎn)。13 d前無誘因出現(xiàn)抽搐,抽搐表現(xiàn)為意識喪失、呼之不應(yīng)、雙眼上翻凝視,四肢強(qiáng)直抖動,無大小便失禁、尖叫、口唇青紫、嘴角歪斜、口吐白沫等;持續(xù)約10~30 s后自行緩解,緩解后疲倦、乏力;期間抽搐約7~8次/d;抽搐后出現(xiàn)發(fā)育倒退明顯,幾乎不發(fā)音(偶發(fā)“ba”音),運(yùn)動倒退為不能抬頭。查體:體質(zhì)量15 kg,身長94 cm,頭圍46.5 cm。四肢肌力:Ⅴ級,四肢肌張力稍低,雙膝反射,腹壁反射未引出,雙巴士征陰性。雙足小,雙足下垂,右足長度比左足短約0.5 cm。手小。脊柱無彎曲。視聽可。眼神交流少。出生史:足月社會因素剖宮產(chǎn),母孕前4月嘔吐嚴(yán)重,出生3.6 kg。既往易磨牙,白天多,睡時少,磨牙聲音大。
1.2 檢查
1歲5月時顱腦MR平掃+DWI:腦室、池、溝增寬(見圖1)。1歲5月時肌電圖:四肢肌廣泛神經(jīng)源性損害。1歲5月時Gesell智能發(fā)育診斷量表:總發(fā)育商(developmental quotient, DQ)26分,其中應(yīng)人能相當(dāng)于3月齡(DQ 18分),應(yīng)物能相當(dāng)于4月齡(DQ 24分),動作能粗大運(yùn)動相當(dāng)于5月齡(DQ 29分),精細(xì)動作相當(dāng)于5月齡(DQ 29分),言語能相當(dāng)于6月齡(DQ 29分)。1歲6月時進(jìn)行性脊肌萎縮癥基因檢測:正常。2歲時尿有機(jī)酸、血氨基酸和酰基肉堿譜無異常。3歲5月時腦電圖示正常。全基因組及線粒體基因檢測:北京康旭醫(yī)學(xué)檢驗(yàn)所檢出患者存在人類基因突變數(shù)據(jù)庫(human genome mutation database, HGMD)已報道過的MECP2基因雜合突變c.316C>T(P.Arg106Trp,見圖2),患者父親母親正常MECP2基因型,患者突變位點(diǎn)改變蛋白質(zhì)甲基CpG結(jié)合域(methyl-CpG binding domain,MBD)中保守的氨基酸,據(jù)美國醫(yī)學(xué)遺傳學(xué)與基因組學(xué)學(xué)會(American College of Medical Genetics and Genomics, ACMG)指南,通過PS1+PS2+PS4+PM1+PM2+PP3證據(jù)證明上述變異為致病,本研究基因突變分析獲貴州醫(yī)科大學(xué)附屬醫(yī)院倫理委員會批準(zhǔn)(批準(zhǔn)號:20101124),并獲患兒監(jiān)護(hù)人的知情同意并簽署知情同意書。3歲8月時視頻腦電圖(VEEG):發(fā)作間期VEEG異常,表現(xiàn)為背景彌漫性混合慢波發(fā)放,前頭部著;雙側(cè)前頭部、中央?yún)^(qū)不規(guī)則尖波、尖慢波、慢波發(fā)放,睡眠期著(見圖3);監(jiān)測到醒睡各期3次強(qiáng)直陣攣發(fā)作,清醒期2次,睡眠期1次,表現(xiàn)為睜眼、凝視→四肢強(qiáng)直→四肢節(jié)律性抖動,同期EEG為廣泛性低-中波幅快波、尖波節(jié)律→高-極高波幅不規(guī)則棘波、棘慢波、慢波節(jié)律,伴持續(xù)肌電爆發(fā)→波幅漸高、節(jié)律漸慢,伴雙上肢節(jié)律性肌電爆發(fā),持續(xù)約44~48 s,發(fā)作期VEEG見圖4。
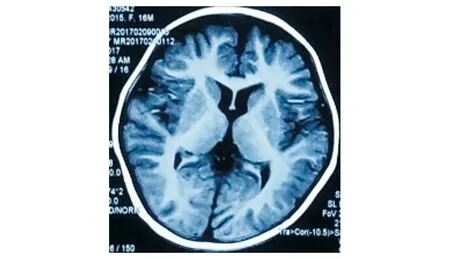
圖1 患者1歲5月時腦MR T1像
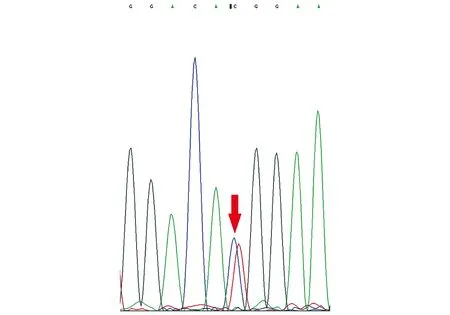
箭頭標(biāo)出雜合突變位點(diǎn):MECP2 c.316C>T(P.Arg106Trp)圖2 患者基因檢測結(jié)果
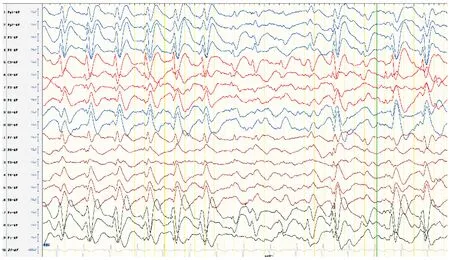
圖3 患者3歲8月時發(fā)作間期VEEG
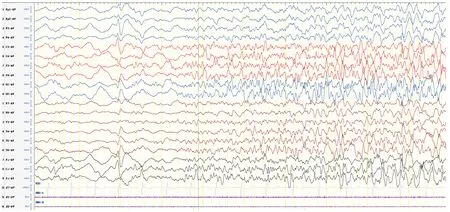
圖4 患者3歲8月時發(fā)作期VEEG
1.3 診斷、治療和隨訪
患者最終診斷:Rett綜合征;癲癇:①強(qiáng)直-陣攣發(fā)作,②MECP2相關(guān)發(fā)育性癲癇性腦病。治療:入院予丙戊酸鈉21.3 mg/(kg·d)口服治療4 d后癲癇發(fā)作控制,但仍有明顯發(fā)育倒退,故排除禁忌后調(diào)整治療為開始加KD治療,KD脂肪:(碳水化合物+蛋白質(zhì))的質(zhì)量比為2 ∶1,熱卡為1 000 cal/d,血酮波動在2.2~3.2 mmol/L,血糖3.8~4.6 mmol/L,KD治療共3月,智力運(yùn)動發(fā)育有好轉(zhuǎn)。患者KD治療10d后可抬頭,精神狀態(tài)好轉(zhuǎn)。KD治療1月(3歲9月)后抬頭可,坐不穩(wěn),叫爸爸清楚,眼神交流可,肢體溫度對稱。患者4歲9月時語言維持同前,精神狀態(tài)可,運(yùn)動改善,抬頭穩(wěn),可扶站,坐時前后搖晃,手抖,易張開雙臂運(yùn)動,踝陣攣陽性。患者現(xiàn)在6歲,體質(zhì)量19 kg,身高108 cm,頭圍51 cm。右足溫度低,右腳長度12.5 cm,左腳長度13 cm。膝反射亢進(jìn),踝陣攣陽性,巴氏征陰性。現(xiàn)抬頭穩(wěn),可靠坐,可扶站,余同4歲9月時發(fā)育情況。已經(jīng)2年多無抽搐,VEEG進(jìn)行性好轉(zhuǎn)。患者4歲9月時復(fù)查VEEG:背景慢化;雙側(cè)中央、頂、左側(cè)枕、中后顳區(qū)尖慢波發(fā)放(見圖5)。患者5歲11月時復(fù)查VEEG:背景慢化;左側(cè)Rolandic區(qū)尖波、尖慢波發(fā)放,右側(cè)少見(見圖6)。
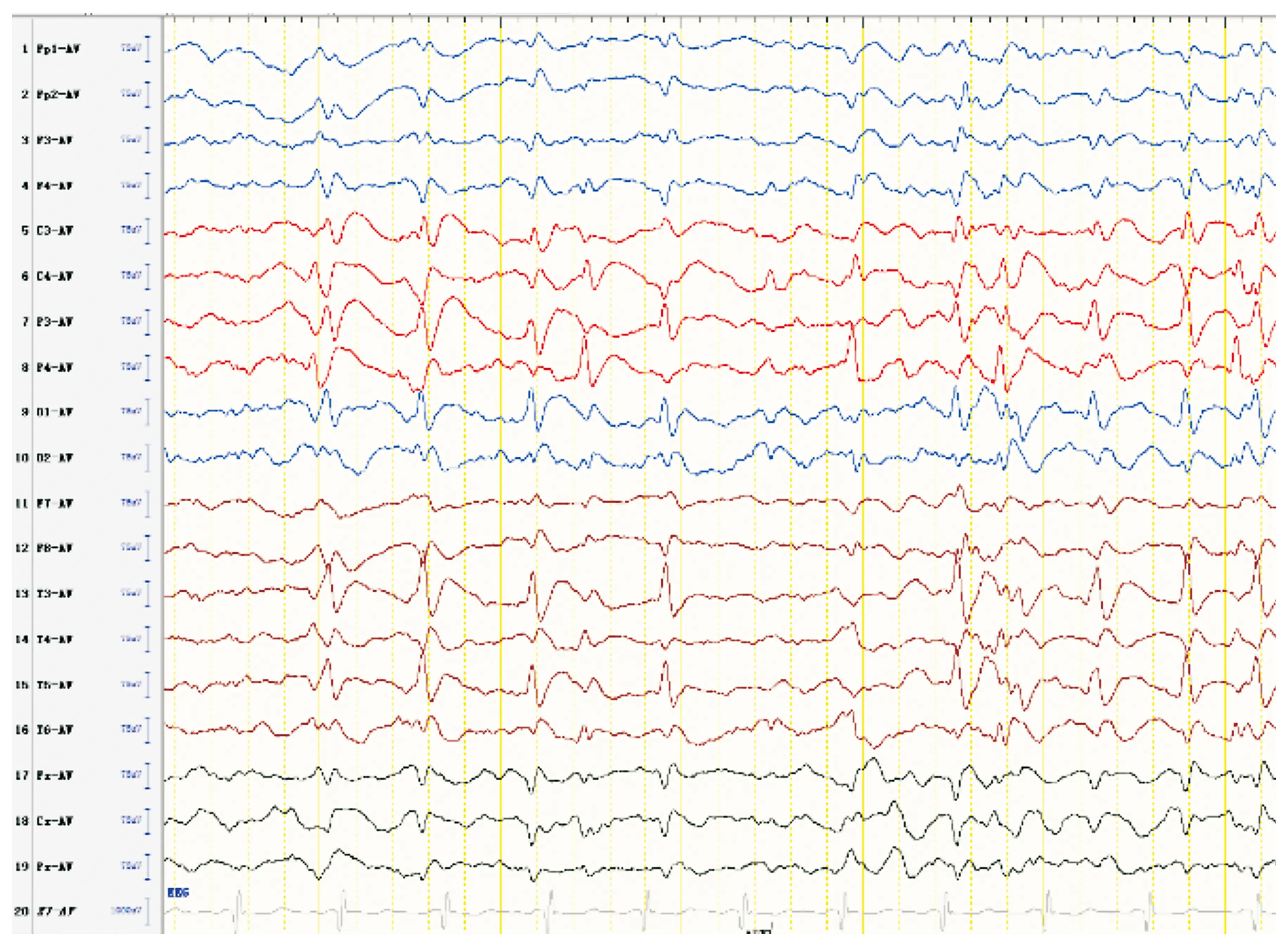
圖5 患者4歲9月時VEEG
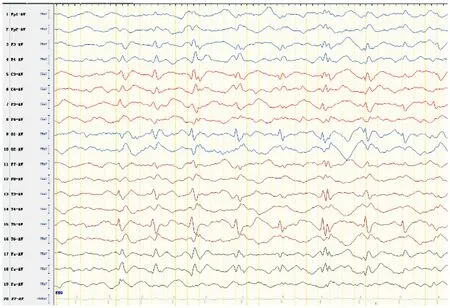
圖6 患者5歲11月時VEEG
2 討論
依據(jù)RTT國際臨床診斷標(biāo)準(zhǔn)[6]和基因檢測結(jié)果,本文報道的患者可以診斷為RTT,結(jié)合有抽搐發(fā)作和VEEG結(jié)果診斷癲癇。根據(jù)發(fā)作類型診斷強(qiáng)直-陣攣發(fā)作。結(jié)合發(fā)育史與癲癇及MECP2基因突變診斷MECP2相關(guān)發(fā)育性癲癇性腦病。RTT世界范圍內(nèi)現(xiàn)沒有最終的治愈方法,僅僅對癥治療,目前研究的RTT基因治療也存在巨大挑戰(zhàn),因MECP2基因治療必須在身體每個細(xì)胞中提供精確劑量的MECP2蛋白,如過多或過少均可使異常MECP2蛋白觸發(fā)RTT樣癥狀[7]。
RTT患者腦內(nèi)γ-氨基丁酸受體(γ-aminobutyric acid receptor, GABAR)信號轉(zhuǎn)導(dǎo)功能失調(diào)[8],GABA是大腦中主要的抑制性神經(jīng)遞質(zhì),GABA能系統(tǒng)異常在RTT癥狀學(xué)中已被證實(shí):僅在GABA能神經(jīng)元Mecp2缺乏的小鼠中可迅速發(fā)展成前爪刻板運(yùn)動,強(qiáng)迫性梳理,學(xué)習(xí)和記憶缺陷,異常社會行為,腦電圖高興奮性,缺乏運(yùn)動協(xié)調(diào),嚴(yán)重呼吸節(jié)律紊亂和過早致死[9]。在Mecp2突變小鼠海馬切片行膜片鉗記錄顯示CA3神經(jīng)元的海馬回路基礎(chǔ)抑制性節(jié)律性活動減少,從而使該回路易受高興奮性影響[10]。已知腦源性神經(jīng)營養(yǎng)因子(brain-derived neurotrophic factor, BDNF)可作為一種有效的調(diào)節(jié)劑,在神經(jīng)元發(fā)育的多個方面以及突觸傳遞和可塑性方面發(fā)揮作用,有研究表明RTT中有BDNF表達(dá)受損[11,12]。
我們使用作用于GABA信號系統(tǒng)的抗癲癇發(fā)作藥丙戊酸鈉治療RTT的癲癇發(fā)作。患者癲癇發(fā)作很快控制,但仍有明顯發(fā)育倒退,因KD除了有廣泛的抗驚厥作用外,還可改善癲癇患者的認(rèn)知/精神問題,且可改善癲癇患者運(yùn)動功能和日常生活活動能力,故本患者開始加用KD治療。
既往研究表明KD有廣泛的抗驚厥作用,機(jī)制有生成酮體和多不飽和脂肪酸;谷氨酸脫羧酶激活,誘導(dǎo)GABA合成;增加大腦中抑制性神經(jīng)遞質(zhì)胍丁胺;激活過氧化物酶體增殖物激活受體,調(diào)節(jié)抗炎、抗氧化和線粒體基因;上調(diào)鈣結(jié)合蛋白緩沖胞內(nèi)鈣的能力具有神經(jīng)保護(hù)潛力;介導(dǎo)抑制凋亡因子神經(jīng)保護(hù);改變腸道微生物等抗驚厥[13]。KD還可以改善癲癇患者的認(rèn)知/精神問題,主要表現(xiàn)在注意和警覺領(lǐng)域,顯著改善視覺注意力。機(jī)制有抗氧化應(yīng)激;維持能量供應(yīng);調(diào)節(jié)脫乙酰化活性和炎癥反應(yīng)等[14]。且KD可改善癲癇患者運(yùn)動功能和日常生活活動能力,包括躺、翻滾、坐、爬、跪、站、走、跑、跳等。機(jī)制有改善突觸可塑性;增加運(yùn)動和體感皮層區(qū)域多巴胺代謝產(chǎn)物等[15]。
KD在RTT中主要通過GABA能和谷氨酸能信號系統(tǒng)有效地治療驚厥等RTT癥狀[16];KD也可以通過BDNF介導(dǎo)的作用或通過增加腺苷產(chǎn)生改善RTT癥狀[11,12,16,17]。Haas等[18]1986年報道了5名RTT,KD改善了癲癇發(fā)作,略微改善了行為,運(yùn)動技能和體重增加。Liebhaber等[19]2003年報告了1例RTT伴難治性癲癇,8歲時開始KD治療4年,發(fā)作頻率降低70%,顯著改善注意力、接觸交往、行為和語言(講更長的句子,小故事;說話更多和更清楚)。Giampietro等[20]2006年報告了1例女性非典型RTT變異患者,KD顯著減少癲癇發(fā)作。Carabalo等[21]2011年報告了1例難治性癲癇的RTT患者,KD減少癲癇發(fā)作75%~99%。我們報告的RTT患者,丙戊酸鈉可有效治療其癲癇發(fā)作,添加KD治療后,患者在運(yùn)動、智力方面有一定改善。
總結(jié),RTT患者需要綜合治療,雖然本例患者KD治療時間相對較短,但KD對RTT的運(yùn)動、智力方面有一定改善,為RTT治療提供了一定的治療希望,但更深入的治療需要進(jìn)一步研究。KD對RTT有積極作用,是可供選擇的一種治療方案。
猜你喜歡
中國民間療法(2021年5期)2021-06-09 09:21:04
中華養(yǎng)生保健(2020年2期)2020-11-16 00:49:00
解放軍醫(yī)學(xué)院學(xué)報(2020年12期)2020-03-29 05:11:46
中成藥(2017年6期)2017-06-13 07:30:35
飲食科學(xué)(2017年5期)2017-05-20 17:11:53
臨床醫(yī)藥文獻(xiàn)雜志(電子版)(2017年11期)2017-05-17 04:48:10
安徽醫(yī)科大學(xué)學(xué)報(2015年9期)2015-12-16 11:09:44
中國當(dāng)代醫(yī)藥(2015年7期)2015-03-01 02:01:13
西南軍醫(yī)(2015年4期)2015-01-23 01:19:30
西部中醫(yī)藥(2014年6期)2014-03-11 16:07:47
