高效液相色譜法測定追風(fēng)透骨丸中α-香附酮和香附烯酮的含量
2023-02-02 07:36:28崔業(yè)波張大勇馬曉靜王路宏
中國藥物經(jīng)濟學(xué) 2023年12期
關(guān)鍵詞:方法
宋 瑩 崔業(yè)波 張大勇 馬曉靜 王路宏
追風(fēng)透骨丸由制川烏、白芷、制草烏、香附(制)等20余味中藥材加工制成,具有祛風(fēng)除濕,通經(jīng)活絡(luò),散寒止痛的功效,臨床廣泛用于風(fēng)寒濕痹,肢節(jié)疼痛,肢體麻木[1]。其中,香附(制)在處方中具有行氣止痛的重要作用[2]。追風(fēng)透骨丸質(zhì)量標準現(xiàn)收錄于《衛(wèi)生部藥品標準·中藥成分制劑第十八冊》[3]和《中華人民共和國藥典(2020年版)》[4],上述標準僅將香附(制)列入鑒別項,缺乏對其含量的質(zhì)控指標,可見其質(zhì)量標準并不完善,而本制劑是國家醫(yī)保目錄甲類藥品,臨床治療必需,且使用廣泛。因此,對其質(zhì)量進行全面、嚴格控制,十分必要。本文通過查閱文獻[5-14],建立高效液相色譜法(HPLC)測定香附(制)中α-香附酮和香附烯酮的含量,為追風(fēng)透骨丸中香附(制)提供更全面、可靠的質(zhì)量控制依據(jù)。
1 儀器與試藥
1.1 儀器
1260型安捷倫高效液相色譜儀(安捷倫科技有限公司,配二極管陣列檢測器);BT125D型電子天平(北京賽多利斯儀器有限公司,感量:0.01/0.1 mg)。
1.2 試藥與樣品
對照品:α-香附酮對照品(中國食品藥品檢定研究院,含量:99.8%,批號:110748-202117)、香附烯酮對照品(Shanghai Standard Technology Co.,Ltd,批號:ST77690181)。試劑:甲醇(DIKMA,批號:50102)為色譜純,乙酸乙酯(國藥集團化學(xué)試劑有限公司,批號:20200610)為分析純,水為超純水(自制)。樣品:追風(fēng)透骨丸購于市區(qū)零售藥店,來源于同一藥品生產(chǎn)企業(yè),批號:K10061、K12089、P12099,規(guī)格:每10丸重1 g,質(zhì)量執(zhí)行標準:《中華人民共和國藥典》。
2 方法與結(jié)果
2.1 色譜條件
色譜柱為ZORBAX SB-C18(4.6×250 mm,5 μm);以甲醇(A)-水(B)(65∶35)為流動相;流速為1.0 ml/min;柱溫為30 ℃;進樣量:5 μl;檢測波長:254 mn。
2.2 溶液制備
2.2.1 對照品混合溶液的制備精密稱取α-香附酮20.09 mg,置50 ml棕色容量瓶中,香附烯酮10.56 mg,置20 ml棕色容量瓶中,分別加甲醇溶解并定容至刻度,搖勻,再分別精密量取上述α-香附酮對照品溶液2 ml、香附烯酮對照品溶液4 ml,置同一10 ml棕色容量瓶中,用甲醇定容至刻度,制成每毫升含α-香附酮80.36 μg、香附烯酮211.2 μg的混合溶液。
2.2.2 供試品溶液的制備精密稱取追風(fēng)透骨丸細粉約20 g,置適宜圓底燒瓶中,加水200 ml,連接揮發(fā)油測定器,在揮發(fā)油測定器上端加水使充滿其刻度部分,至有水流入燒瓶時為止,再加入乙酸乙酯1 ml,連接回流冷凝管,將圓底燒瓶置電熱套中緩慢加熱至沸,并保持微沸約1.5 h,停止加熱,分取乙酸乙酯層,置10 ml棕色容量瓶中,再用少量甲醇沖洗冷凝管,將沖洗液并入上述10 ml棕色容量瓶中,加甲醇定容至刻度,搖勻,用0.45 μm濾膜濾過,即得。
2.3 方法學(xué)考察
2.3.1 專屬性試驗分別精密吸取“2.2”項下對照品混合溶液、供試品溶液及陰性對照溶液[按照處方比例制備缺香附(制)的陰性樣品,按“2.2.2”供試品溶液制備方法制成陰性對照溶液]各5 μl,按“2.1”項下色譜條件進樣測定,記錄色譜圖。結(jié)果供試品溶液在與對照品混合溶液色譜圖相同位置處,出現(xiàn)保留時間一致的色譜峰,且與相鄰色譜峰分離完全,分離度均大于1.5,符合要求,α-香附酮和香附烯酮理論板數(shù)均不低于8 000。陰性對照溶液色譜圖中,未出現(xiàn)待測成分色譜峰,說明本方法專屬性良好。見圖1~3。
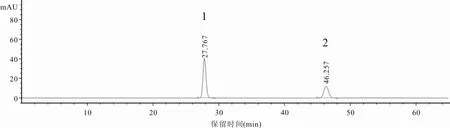
圖1 α-香附酮和香附烯酮對照品混合溶液HPLC色譜
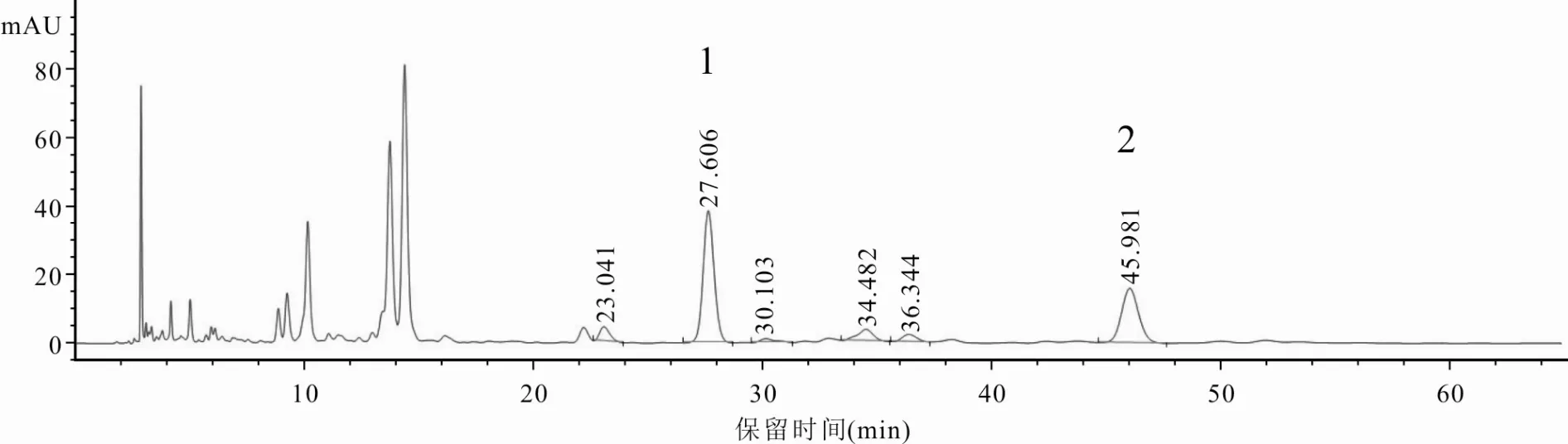
圖2 追風(fēng)透骨丸供試品溶液HPLC色譜
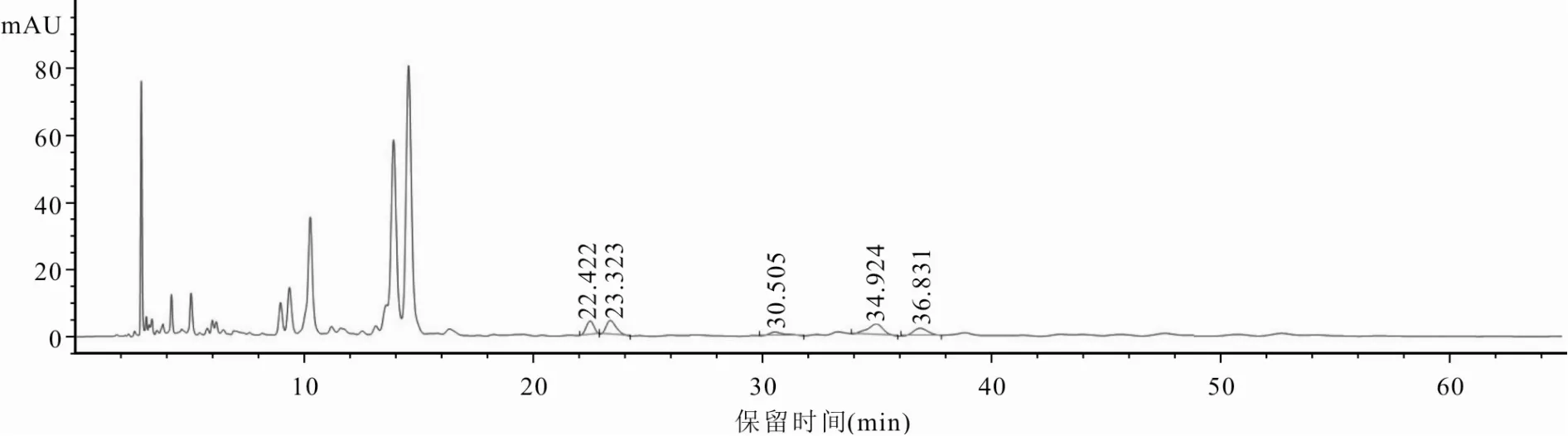
圖3 追風(fēng)透骨丸陰性對照溶液HPLC色譜
2.3.2 線性關(guān)系精密量取“2.2.1”項下對照品混合溶液1、2、5、8、10 μl注入液相色譜儀,按“2.1”項下色譜條件進樣測定,記錄峰面積。以待測成分進樣質(zhì)量為橫坐標(X)、峰面積為縱坐標(Y),進行線性回歸,結(jié)果表明兩種待測成分在各自檢測質(zhì)量范圍內(nèi),線性關(guān)系良好。見表1。
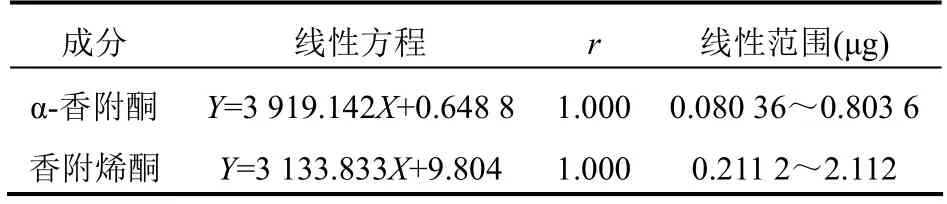
表1 追風(fēng)透骨丸中α-香附酮和香附烯酮標準曲線測定結(jié)果
2.3.3 精密度試驗精密量取同一對照品混合溶液5 μl,注入液相色譜儀中,按“2.1”項下色譜條件,連續(xù)進樣分析6次,結(jié)果:α-香附酮、香附烯酮峰面積的相對標準偏差(RSD)分別為0.40%、0.45%,表明儀器精密度良好。
2.3.4 重復(fù)性試驗取同一批追風(fēng)透骨丸(批號:K10061)細粉約20 g,按“2.2.2”項下方法制備供試品溶液6份,同法測定,結(jié)果供試品中每克含香附烯酮0.041 mg,RSD值為1.0%,含α-香附酮0.021 mg,RSD為1.4%,說明該方法重復(fù)性良好。
2.3.5 穩(wěn)定性試驗取同一批追風(fēng)透骨丸(批號:K10061)供試品溶液,分別在0、2、4、6、8、10、12、24 h時,精密量取5 μl,注入液相色譜儀中,按“2.1”項下色譜條件分析,分別記錄待測成分峰面積,其24 h內(nèi)的RSD分別為:0.6%、0.4%(n均等于8),表明供試品溶液在24 h內(nèi)穩(wěn)定。
2.3.6 回收率試驗精密稱取已知含量(香附烯酮0.041 mg/g、α-香附酮0.021 mg/g)追風(fēng)透骨丸(批號:K10061)細粉約10 g,再分別加入濃度為2.074 mg/ml的香附烯酮對照品溶液0.2 ml和濃度為1.330 mg/ml的α-香附酮對照品溶液0.2 ml,按“2.2.2”項下方法制備供試品溶液6份,同法測定,計算各樣品中香附烯酮和α-香附酮的加樣回收率。見表2。
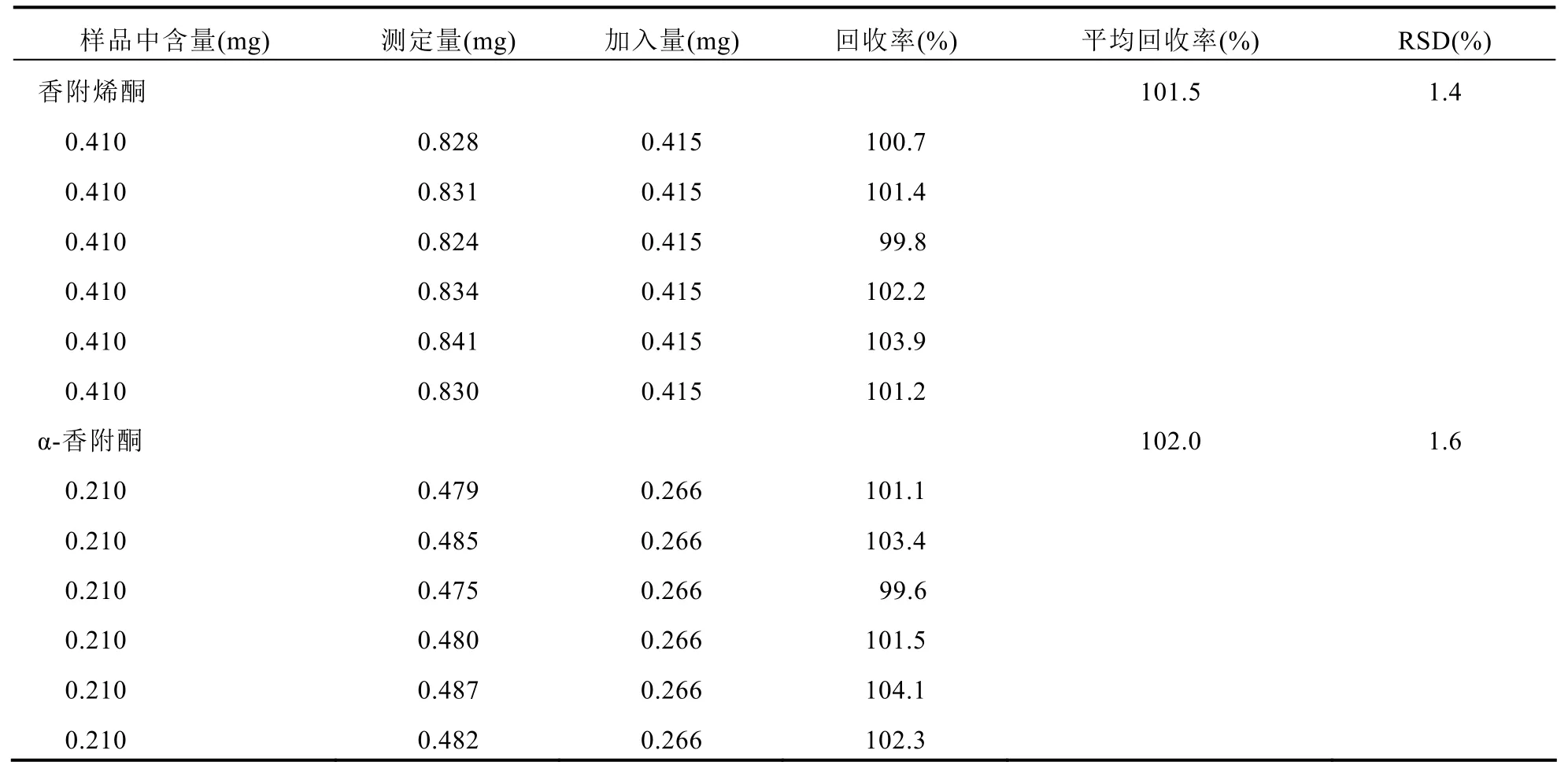
表2 樣品中香附烯酮和α-香附酮加樣回收率試驗結(jié)果
2.4 樣品的含量測定
分別對3批追風(fēng)透骨丸中香附烯酮和α-香附酮的含量進行測定。見表3。
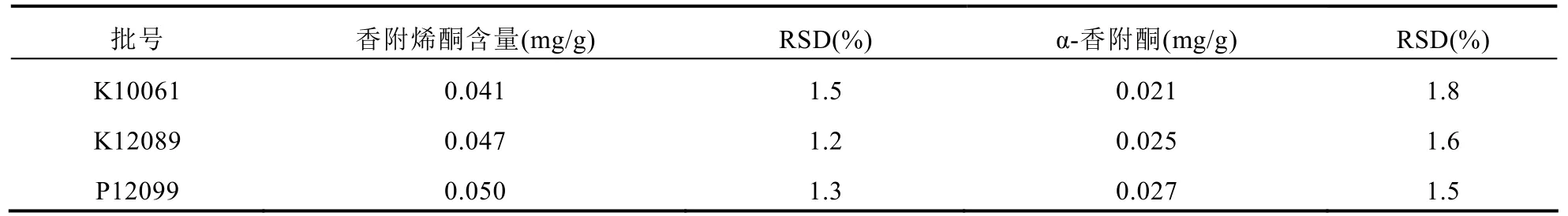
表3 追風(fēng)透骨丸中α-香附酮和香附烯酮含量測定結(jié)果
3 討論
3.1 供試品待測成分制備方法的選取
本文考慮到追風(fēng)透骨丸為中藥成方制劑,由20余種中藥材原粉直接入藥制成,成分復(fù)雜,采用超聲、加熱回流直接提取的方法,得到的供試品溶液成分較多,色譜峰較難分離,而α-香附酮和香附烯酮均為香附(制)中具有揮發(fā)性的揮發(fā)油成分,故本文采用水蒸氣蒸餾法對其進行提取,既符合待測成分本身的理化性質(zhì),同時又起到純化待測成分的作用,結(jié)果表明該提取方法得到的供試品液相色譜圖,雜質(zhì)峰較少,待測成分色譜峰宜與相鄰色譜峰分離,且分離度滿足要求。
3.2 不同色譜柱的考察
本文分別采用美國安捷倫科技有限公司Agilent TC-C18、ZORBAX SB-C18和北京迪馬科技有限公司Dimaonsil C18色譜柱(250×4.6 mm,5 μm),對建立的檢測方法中色譜柱選用進行考察,結(jié)果用以上3種色譜柱得到的色譜圖中,待測成分色譜峰與相鄰色譜峰分離度均大于1.5,且理論塔板數(shù)均能達到8 000以上,符合要求,表明以上色譜柱均可用于該方法的檢測,本文選用以上3種色譜柱中較為常用的ZORBAX SB-C18柱進行試驗。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學(xué)生數(shù)理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
