抗Mi-2抗體陽性特發性炎性肌病臨床及病理特征分析:附7例報道及文獻復習
2023-02-04 02:49:40藺穎陳娟劉孟洋石強
疑難病雜志 2023年1期
藺穎,陳娟,劉孟洋,石強
特發性炎性肌病(idiopathic inflammatory muscle disease,IIM)是一組異質性的自身免疫性疾病,以進行性肌無力、肌電圖異常、肌酶升高和肌肉炎性細胞浸潤為特征,常伴有其他系統受累表現。2018年歐洲神經肌肉中心(European Neuromuscular Centre ,ENMC)基于臨床表現、骨骼肌特征性病理改變及肌炎特異性自身抗體(myositis specific autoantibodies, MSAs)將IIM分為4型:多發性肌炎、皮肌炎(dermatomyositis,DM)、散發性包涵體肌炎和壞死性自身免疫性肌炎[1]。抗染色體重塑酶(nucleosome-remodeling deacetylase complex, Mi-2)抗體是迄今已鑒定出的5種皮肌炎特異性抗體(dermatomyositis specific antibody,DMSA)之一[2]。本研究應用免疫印跡法篩選抗Mi-2抗體陽性IIM患者,歸納其臨床及骨骼肌病理特點,并結合文獻報道進行比較,以提高對此類患者的認識,報道如下。
1 臨床資料
1.1 一般資料 收集2019年1月1日—2022年2月28日在解放軍總醫院第一醫學中心行肌肉活檢篩選出抗Mi-2抗體陽性IIM患者7例臨床資料。7例患者中男2例,女5例,就診年齡21~70歲,起病年齡21~69歲;慢性病程5例,急性起病2例。腫瘤和其他免疫系統疾病:甲狀腺低度惡性癌、系統性紅斑狼瘡(SLE)病史、陰莖癌手術史各1例;甲狀腺功能減退病史2例,其中1例在疫苗接種后發病。本研究經醫院倫理委員會批準(S2022-399-01),所有患者及家屬均知情同意并簽署知情同意書。
1.2 臨床表現 7例患者中肌痛5例,肌萎縮3例;頸部肌無力2例,近端肌無力6例(85.71%),球肌受累2例,呼吸肌受累1例,不伴肌無力1例。肌電圖表現為運動單位電位時限明顯縮短,平均電壓及重收縮時峰值電壓降低,提示肌源性損害。
肌外表現:(1)皮疹:4例出現皮疹,受累范圍廣泛,均有面部受損,頸部和手指受累2例,軀干部(胸、腹、背)和四肢受累2例,皮損為多發片狀深暗紅色,粗糙、表面有鱗屑,水皰、血皰、破潰后結痂或形成潰瘍,伴瘙癢、疼痛。(2)呼吸系統損害:合并肺部感染4例,合并間質性肺病(interstitial lung disease,ILD)1例。(3)關節痛:合并關節痛3例。
1.3 實驗室檢測 血清肌酸激酶(creatine kinase,CK)峰值波動范圍15.6~14 240 U/L。WBC升高1例。1例 ANA滴度1∶100,其余病例ANA滴度均≥1∶1 000。MSAs: 抗Mi-2抗體均陽性,Mi-2ɑ的波動范圍(-~+++),Mi-2β的波動范圍(-~++),其中Mi-2ɑ(+++)單陽性1例,Mi-2β(+)單陽性3例,Mi-2ɑ和Mi-2β聯合陽性2例,Mi-2陽性但未區分亞型1例。1例伴抗PM/SCL抗體(+)、抗SRP抗體(+)、抗Jo-1抗體(+);1例伴抗Ku抗體(+);1例伴Ro-52(++)、U1-nRNP(+)。
1.4 影像學檢查 胸部CT:5例患者可見肺葉的炎性改變,其中1例可見雙肺外周間質性肺炎,雙側胸膜增厚,心包少量積液。4例可見散在小結節,均考慮為良性。
MR表現:2例完成骨骼肌MR檢查,分別可見雙側骨盆及大腿肌群、頸項部肌肉及棘間韌帶,片狀長T2高信號和STIR高信號,提示肌肉和韌帶炎性改變,見圖1。
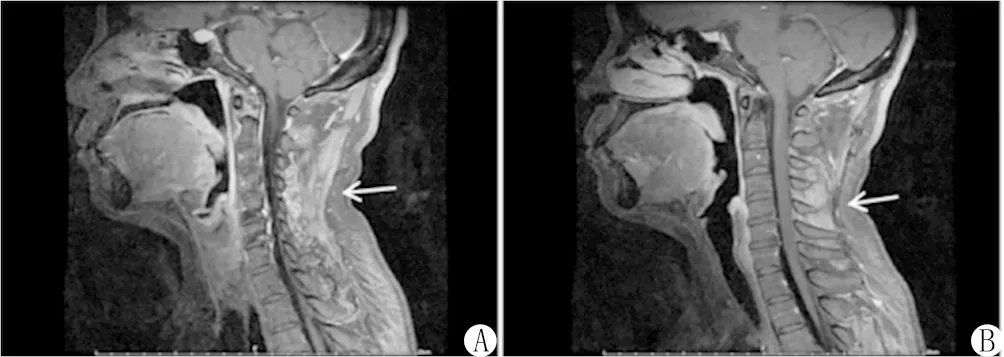
注:A、B均為 T2圖像,分別可見頸后肌群及棘間韌帶片狀長T2高信號,提示肌肉和韌帶的炎性改變
1.5 骨骼肌病理結果 7例骨骼肌病理可見炎性肌病的病理改變,符合束周免疫肌病的病理特點。具體可見束周肌纖維變性、萎縮和壞死,并伴炎性細胞浸潤;可見肌纖維片狀分布的中央壞死及周圍再生肌纖維(centrally necrotic-and-peripherally regenerating ,CNPR)。免疫組織化學染色MHC-Ⅰ肌內衣表達陽性和MxA束周肌纖維膜和肌漿陽性表達,見圖2。
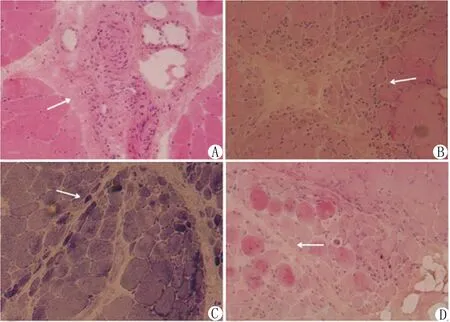
注:A.肌間質血管周圍大量炎性細胞浸潤(HE染色,×200);B.束周肌纖維變性萎縮伴炎性細胞浸潤(HE染色,×200);C.可見束周肌纖維變性萎縮壞死(NADH染色,×200);D.肌纖維微梗死和中央壞死周圍再生肌纖維(HE染色,×200)
1.6 治療與轉歸 7例患者使用糖皮質激素和免疫抑制劑(包括嗎替麥考酚酯3例、環磷酰胺2例、硫唑嘌呤1例、他克莫司1例)聯合用藥,治療后3個月,患者病情均較前好轉,表現為皮疹范圍的縮小和肌無力癥狀的好轉,但需長期隨訪有無惡性腫瘤。
2 討 論
DMSAs的發現改變了基于臨床癥狀(1975年Bohan和Peter基于皮膚損傷和肌肉無力)和臨床病理學標準(2018ENMC-DM)提出的DM分類標準[1]。在2018年ENMC-DM共識中,將以下5個DMSAs作為DM血清學標準[2]:抗轉錄中介因子1-γ、抗Mi-2、抗黑色素瘤分化基因5(melanoma differentiation antigen 5,MDA5)、抗核基質蛋白2和抗小泛素樣修飾激活酶。DMSA相關臨床表型和病理表型研究大多數僅限于小型研究,并不包括在2018年的ENMC-DM中。
盡管抗Mi-2抗體是IIM中發現的第一個自身抗體,但抗Mi-2抗體陽性IIM患者的臨床及病理特征仍未明確[3]。1976年Reichlin等[4]在1例名為“Mi”的DM患者血清中發現一種新的抗體,并命名為抗Mi-2抗體;1980年Nishikai等[5]將Mi抗體分為抗Mi-1抗體和抗Mi-2抗體,其中抗Mi-2抗體屬于MSAs;1995年Ge等[6]通過噬菌體展示技術發現240 kD條帶上的主要靶抗原分別為Mi-2ɑ和Mi-2β,對應抗Mi-2ɑ抗體和Mi-2β抗體。本研究收集7例肌肉活檢病例,抗Mi-2抗體陽性但未區分亞型1例,抗Mi-2ɑ和Mi-2β抗體雙陽性2例,抗Mi-2ɑ抗體單陽性1例,抗Mi-2β抗體單陽性3例。文獻報道抗Mi-2抗體可與其他MSAs同時出現,如抗NXP2和Jo-1抗體[7]。本研究觀察到1例同時出現MSAs(SRP、Jo-1)和肌炎相關性自身抗體(MAAs)(PM/Scl),2例伴發MAAs(Ku、Ro52/U1-nRNP),上述病例骨骼肌病理符合DM的改變,故仍歸類為抗Mi-2抗體陽性IIM。
既往有多個團隊對于抗Mi-2抗體陽性的IIM的發病情況進行統計研究。Betteridge等[8]于2019年的一項歐洲IIM的聯合隊列研究,其中抗Mi-2抗體陽性的IIM患者發病率為5.4%(88/1 637)。Li等[9]于2019年對497例IIM患者進行了回顧性研究,其中抗Mi-2抗體陽性的發病率為6.2%(31/497)。Gómez等[10]于2021年的一項基于IIM患者的多中心橫斷面研究,其中抗Mi-2抗體陽性的IIM患者的發病率為10.2%(23/226),其中女性占比為69.57%(16/23),發病年齡約為43.4±17.6歲,其中為慢性病程者占比39.13%(9/23)。抗Mi-2抗體陽性的IIM患者較為少見,發病人群主要為中年人,男女均可受累,可表現為慢性病程。本組7例患者的發病年齡為43.7±16.3歲,女性占比為71%(5/7),慢性病程占比為71%(5/7),除本組患者慢性病程人數較多外,其余均與文獻報道的范圍大致相符。
抗Mi-2抗體陽性IIM的臨床表型包括高CK、經典DM皮疹和肌無力[3,10-13]。Sharma等[14]納入34例幼年型皮肌炎(JDM)患兒僅有1例抗Mi-2抗體陽性,在入組時肌肉力量正常。抗Mi-2抗體陽性的IIM患者表現為典型的皮損和肌炎,很少與內部惡性腫瘤和ILD相關。Hengstman等[15]系統評價來自歐洲6個中心的48例抗Mi-2 IIM陽性肌炎患者中,伴關節痛39例,伴關節炎26例,伴雷諾現象25例,伴ILD 16例。Hydzik等[16]報道1例抗Mi-2抗體相關IIM患者出現經典皮損、關節痛、肌痛和肌酶升高。本組7例患者中,CK最高14 240 U/L,2例肌酶正常,4例伴面部、頸部、手指甚至軀干皮膚紅斑疹。6例近端肌無力,1例肌力正常,伴吞咽困難2例,伴關節痛3例,伴肌痛5例,伴肌萎縮3例。Hamaguchi等[17]研究376例日本成年IIM患者中9例抗Mi-2陽性患者與無ILD或惡性腫瘤的經典DM相關。Li等[9]入組497例IIM患者中發現31例抗Mi-2抗體陽性,其中22例進行胸部CT檢查,診斷伴ILD者4例,不伴ILD者18例,經Logistic回歸分析發現,抗Mi-2抗體是ILD發生的保護因素之一。抗Mi-2α抗體和抗Mi-2β抗體陽性IIM中ILD和惡性腫瘤的發生率較低[18-19]。本組7例病例中,僅1例伴發ILD,2例伴發癌癥,SLE、甲狀腺功能減退/疫苗接種后和甲狀腺功能減退的免疫性疾病各有1例。雖吻合文獻中報道的ILD患病率低,但惡性腫瘤發生率仍較高。
在2018年的ENMC-DM中,只有束周萎縮和MxA的肌纖維表達被作為明確的病理標準。肌漿MxA表達比束周萎縮對DM診斷的敏感度更高[20-23]。但肌漿MxA表達可以分布束周肌纖維,也可單純散在或彌漫分布,束周分布常見于抗Mi-2 DM,單純散在或彌漫分布常見于抗MDA5 DM[24]。抗Mi-2抗體陽性IIM的組織學特征符合肌束周免疫肌病(immune myopathies with perimysial pathology,IMPP)[25],如束周萎縮、束周壞死、束周炎性反應和束周MxA表達。本研究中病例同樣出現束周炎性反應、束周萎縮、束周壞死和束周肌漿MxA表達,符合抗Mi-2抗體陽性IIM的肌束周病理特點。CNPR經常存在于微梗死的肌肉活檢中,卻與微梗死無關,常見于抗Mi-2抗體陽性IIM中,可能是通過一種不同于抗NXP-2DM的潛在病理機制發展起來[24]。本研究中可見CNPR樣病理改變,符合文獻報道。
抗Mi-2抗體陽性IIM對于類固醇的治療反應和預后良好[26]。Deakin等[27]報道抗Mi-2似乎對JDM患者有保護作用,這些患者隨著時間的推移,繼續接受治療的幾率低了7.06倍(95%CI1.41~35.36,P=0.018)。本組7例患者經激素聯合免疫抑制劑治療后,臨床癥狀改善較好,但長期預后仍需進一步隨訪。
總之,抗Mi-2抗體陽性IIM臨床表型多見經典DM紅疹,肌肉活檢可見典型DM束周病理免疫肌病的改變,伴ILD較少,可伴惡性腫瘤或甲狀腺功能減退等免疫相關因素,類固醇聯合免疫抑制劑反應較好,但長期預后尤其伴發惡性腫瘤的患者需進一步的隨訪。
利益沖突聲明:所有作者聲明無利益沖突
作者貢獻聲明
藺穎:資料收集整理,論文撰寫;陳娟、石強:設計研究方案,論文審核;劉孟洋:資料收集整理
