WO3/BiOCl0.7I0.3光催化劑的制備及其光催化降解機理
2023-03-01 07:40:10劉海成孟無霜黃哲尤雨花瑞琪曹夢茹
化工進展 2023年1期
關鍵詞:復合材料
劉海成,孟無霜,黃哲,尤雨,花瑞琪,曹夢茹
(蘇州科技大學環境科學與工程學院,江蘇 蘇州 215009)
抗生素自發現以來,因其優越的抗菌性能被廣泛用于制藥、醫療和畜牧業等領域,其中四環素類抗生素(tetracyclines)是抗生素中應用最為廣泛的一類[1?2]。抗生素具有持久性,且難以被生物體完全代謝,通過各種途徑進入水體在水環境中富集,會導致微生物產生抗藥基因[3?4]。抗生素耐藥性引發的細菌感染將嚴重威脅人類的生存安全[5],水體中抗生素的去除問題亟待解決。
傳統水處理技術(生物處理、吸附去除等)在面對抗生素類廢水的處理時,處理能力表現出不足。相較于此,光催化技術因環保綠色、成本低廉和高效降解有機污染物等優勢,成為研究熱點[6?7]。WO3是一種能夠可見光響應的光催化劑,禁帶寬度為2.4~2.8eV,因獨特的晶格結構,具有良好的電子傳輸能力,性質穩定且易于制備,既可用作主催化劑,也可用作助催化劑[8]。但WO3的導帶電位比電子還原氧的電位低,無法將吸附在其表面的氧還原為超氧自由基,電子無法及時清除,導致了較高的電子空穴復合率,大幅降低了光催化性能。有研究證實,可以通過復合半導體構建異質結構的方式來解決這一問題[9]。
異質結構可以優化電子轉移路徑,促進光生電子(e?)和空穴(h+)的分離,從而提高材料光催化性能。Bi等[10]采用水熱法和電沉積法成功制備了在可見光激發下具有優異光催化活性的BiOI/WO3異質結構光催化劑。BiOI禁帶寬度(1.8eV)較窄,對可見光有較好的吸收性能,BiOCl 獨特的層狀結構形成的內部靜電場可以促進電子空穴對的分離,兩者結構相似,且結合在一起可以形成光催化性能更優異的固溶體BiOClxI1?x。如Deng等[11]通過溶劑熱法制備了BiOCl0.7I0.3固溶體,且具有良好的光催化活性。Ma 等[12]采用沉淀法合成了新型BiOCl0.9I0.1/β?Bi2O3復合材料,有效促進了光催化反應中的電荷分離,提高了光催化性能。
因此,可通過簡單的水浴加熱和原位沉淀法將BiOCl0.7I0.3和WO3結合,制備復合光催化劑,并應用于鹽酸四環素的降解研究。復合材料形成異質結構,可以抑制電子空穴對復合速率,拓寬可見光吸收范圍,從而提高對鹽酸四環素的光降解性能。通過自由基捕獲試驗和電子順磁共振檢測(ESR)明確光催化反應的活性物質,并分析推測出WO3/BiOCl0.7I0.3復合光催化材料的光催化作用機制。
1 材料與方法
1.1 試劑與儀器
1.1.1 試劑
鎢酸銨[(NH4)10H2W12O42]、五水硝酸鉍[Bi(NO3)3?5H2O]、乙二胺四乙酸二鈉(EDTA?2Na),上海麥克林生化科技有限公司;乙二醇(C2H6O2)、異丙醇(IPA),無錫市晶科化工有限公司;碘化鉀(KI)、氯化鉀(KCl)、對苯醌(BQ),上海阿拉丁生化科技有限公司。以上試劑均為分析純。鹽酸四環素,純度級別為USP,上海麥克林生化科技有限公司。試驗用水為娃哈哈純凈水。
1.1.2 儀器
掃描電子顯微鏡,Quanta FEG 250 型,美國FEI公司;X射線衍射儀,D8advance型,德國布魯克;紫外可見分光光度計,TU?1900型,北京普析通用儀器有限責任公司;傅里葉紅外光譜儀,Thermo Nicolet Is5型,美國Thermo公司;X射線光電子能譜儀,Escalab 250xi 型,美國Thermo 公司;紫外可見漫反射光譜儀,UV3600 型,日本島津;電子順磁共振光譜儀,Bruker A3000 型,德國布魯克。
1.2 光催化材料的制備
1.2.1 WO3的制備
稱取5g 鎢酸銨固體置于陶制坩堝內,轉移至氣氛爐中以10℃/min 的升溫速度升至520℃,于520℃下煅燒2h,繼續以10℃/min 的升溫速度升溫至540℃煅燒2h。冷卻至室溫后,取出研磨得到淡黃色粉末狀WO3。
1.2.2 WO3/BiOCl0.7I0.3復合材料及BiOCl0.7I0.3的制備
采用水浴加熱和原位沉淀法制備BiOCl0.7I0.3和WO3/BiOCl0.7I0.3。稱取0.01mol 的五水合硝酸鉍加入10mL 的乙二醇中,在90℃水浴條件下攪拌溶解至透明,記為A 溶液。稱取不同物質的量的WO3(0.5mmoL、0.67mmoL、1.0mmoL、2.0mmoL)置于燒杯中,加入10mL 去離子水攪拌后超聲形成分散液,記為B 溶液。稱取7mmoL 氯化鉀和3mmoL 碘化鉀置于燒杯中,加入40mL 去離子水,攪拌至完全溶解,記為C溶液。在90℃水浴條件下,將C液滴加入A 液中,并持續攪拌30min。隨后,在持續攪拌的條件下,將B液滴加入上述溶液中,并于滴加完成后繼續攪拌60min。冷卻至室溫后抽濾,于60℃下真空干燥12h,得到不同摩爾比的BW(20∶1、15∶1、10∶1、5∶1)復合材料,記為BW?x(其中x為BW 復合材料中BiOCl0.7I0.3的含量)。在相同條件下,不添加WO3制備得到BiOCl0.7I0.3單體。
1.3 光催化材料的表征
利用X 射線衍射(X?ray diffraction,XRD)對材料的晶型、物相組成進行分析,掃描區段10°~80°;通過傅里葉變換紅外光譜(Fourier transform infrared spectrometer,FTIR) 分析材料的化學組成、化學鍵;掃描電子顯微鏡(scanning electron microscope,SEM)用于觀察樣品的微觀形貌;X射線 能 譜(X?ray photoelectron spectroscopy,XPS)可以表征樣品的元素組成和價態;紫外可見漫反射光譜(UV?Vis diffuse reflection spectrum,UV?Vis DRS)可以研究樣品的光學吸收性能,并將其與XPS結合,從而對材料禁帶寬度進行分析;電子自旋共振(electron spin?resonance,ESR)用于測定樣品在光催化降解過程中產生的自由基種類。
1.4 光催化降解試驗
以氙燈(300W)模擬可見光源,同時加載420nm 濾光片,以100mL 的鹽酸四環素溶液(20mg/L)為目標污染物,進行光催化降解試驗。將磁性轉子和0.1g光催化材料投加進鹽酸四環素溶液,并轉移到光催化反應裝置中,在黑暗狀態下持續攪拌30min以達吸附平衡。然后打開模擬光源進行光催化反應,在反應期間每隔10min用注射器取3mL 反應樣,隨后經0.22μm 的尼龍濾膜過濾,并將濾液轉移到離心管中待測。采用紫外分光光度法(λ=357nm)對鹽酸四環素溶液樣品進行測定。
1.5 自由基捕獲試驗
以乙二胺四乙酸二鈉(EDTA?2Na)、異丙醇(IPA)、對苯醌(BQ)分別作為空穴(h+)、羥基自由基(?OH)、超氧自由基(?O2?)的捕獲劑[13],在光催化降解鹽酸四環素試驗中,投加催化劑后再分別加入1mmol 的EDTA?2Na、IPA、BQ,其他條件不變,測定在加入不同捕獲劑后溶液中鹽酸四環素濃度隨時間的變化情況。
2 結果與討論
2.1 光催化材料的表征
2.1.1 XRD分析
采用XRD 對材料的晶型和物相結構進行分析(圖1)。在2θ為10°~80°范圍內,制備的WO3樣品在23.143°、23.623°、24.403°、33.285°、34.205°、41.666°、49.967°、55.868°處出現了明顯的衍射峰,分別對應于純WO3的(002)、(020)、(200)、(022)、(202)、(222)、(140)、(420)晶面,這與單斜相WO3(JCPDS No.43?1035)標準卡片基本吻合[14]。將制備的BiOCl0.7I0.3光催化材料的XRD 衍射圖譜與BiOCl (JCPDS No.06?0249) 以 及BiOI (JCPDS No.10?0445) 標準卡片進行對照[15],可以看出BiOCl0.7I0.3樣品在11.417°、25.821°、32.610°、33.259°、40.934°、46.837°、54.216°、58.526°和68.266°處出現了明顯的衍射峰,分別對應(001)、(101)、(110)、(102)、(112)、(200)、(211)、(212)和(220)晶面,說明了BiOCl0.7I0.3固溶體的合成[16]。在BW?15樣品材料的XRD 圖譜中,可以較為明顯地觀察到BiOCl0.7I0.3的衍射峰,且在2θ為23.119°、23.596°、24.373°處可以觀察到WO3的三個優勢衍射峰,分別對應于WO3的(002)、(020)、(200)晶面,但其中WO3的特征衍射峰的強度較弱,推測可能是由于復合材料中WO3的含量較低所致。與此同時,BW?15 圖譜中基本未觀察到雜質峰,表明制備的樣品具有較高的純度。

圖1 WO3、BiOCl0.7I0.3及BW?15光催化劑的XRD圖譜
2.1.2 FTIR分析
對所制備的BiOCl0.7I0.3、WO3和BW?15 光催化材料進行傅里葉紅外光譜分析(圖2)。在波長1382cm?1處明顯觀察到一個強特征峰,這是由Bi—Cl的不對稱伸縮振動產生的[17]。535cm?1處的特征峰由Bi—O伸縮振動產生[18],而位于838cm?1處的特征峰是I—O 伸縮振動產生的[19]。在波數為837cm?1處有一個強特征峰,這是由WO3晶體中O—W—O 鍵的伸縮振動導致的[20],但由于復合材料中WO3的含量較低,因此在BW?15 的譜圖上這個特征峰較為不明顯。以1616cm?1為中心的特征峰是H—O—H鍵的彎曲振動產生的,而以3428cm?1為中心的寬透射帶是由O—H基團的伸縮振動所致,這兩種特征峰由材料的吸附水所產生[21]。以上分析可與XRD 的分析結果相互印證。
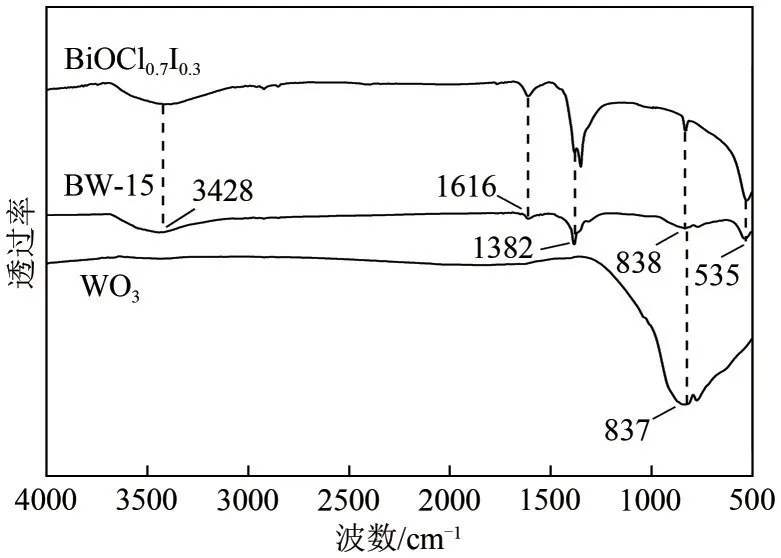
圖2 WO3、BiOCl0.7I0.3及BW?15光催化劑的FTIR圖譜
2.1.3 掃描電子顯微鏡分析
借助掃描電鏡觀察BiOCl0.7I0.3和WO3/BiOCl0.7I0.3復合材料的表面微觀形貌。BiOCl0.7I0.3固溶體在不同放大倍數下呈現出由大量納米薄片組成的花狀[圖3(a)~(c)],尺寸為1~2μm,同時也可以看出沉淀法制備的BiOCl0.7I0.3存在團聚現象,分散性較差。在WO3/BiOCl0.7I0.3復合材料的電鏡圖[圖3(d)]中未觀察到較為明顯的WO3,但觀察到BiOCl0.7I0.3團聚呈大小不一的塊狀分布。由于掃描電鏡只能觀測到材料的表觀形態,而無法觀測到材料的內部形態,且由XRD 和FTIR 的分析結果已經證實了材料中WO3的存在,因此推測可能是因為BiOCl0.7I0.3密集附著生長在了WO3的表面,且復合材料中WO3的含量較少,被BiOCl0.7I0.3掩蓋在了內部所致。

圖3 不同光催化劑的SEM圖譜
2.1.4 X射線能譜分析
通過對BW?15 的全范圍XPS 掃描譜圖[圖4(a)]進行分析,研究了制備的復合光催化材料BW?15的元素組成和化學狀態。由圖中可以清晰地看到Bi、Cl、I、O、W 五種元素均存在于BW?15 復合材料中,這與XRD以及FTIR光譜的顯示結果可以互為佐證。
通過對XPS精細譜進行分析,可以研究光催化材料的各元素價態和成鍵情況。圖4(b)是Bi 4f光譜圖,從圖中可以看出Bi 4f5/2和Bi 4f7/2軌道可以擬合兩個峰,兩個主峰分別對應的結合能為164.90eV和159.60eV,表明材料中的Bi 是以Bi3+的形式存在[22]。圖4(c)是Cl 2p高分辨率光譜,以198.30eV和199.90eV 的結合能為中心的峰分別屬于Cl 2p3/2和Cl 2p1/2自旋軌道能級,表明元素Cl 是以Cl?形式呈現[23]。
O 1s的精細譜如圖4(d)所示,可知O 1s譜圖中有三個擬合峰,分別在532.50eV、531.15eV 和530.25eV結合能處,分別對應于W—O鍵、吸附在樣品表面的—OH和[Bi2O2]2+晶格中的Bi—O鍵[24?25]。

圖4 BW?15復合光催化劑的XPS圖譜
在I 3d的XPS光譜圖[圖4(e)]中可以清晰地觀察到兩個擬合峰,分別位于630.80eV 和619.30eV,這兩個峰是獨立的峰,分別歸屬于I 3d3/2和I 3d5/2自旋軌道,其中630.80eV 對應I3?,619.30eV 對 應I?[25]。W 4f的高分辨率光譜如圖4(f)所示,結合能峰位于29.60eV和36.15eV,結合能峰分別對應W 4f7/2和W 4f5/2自旋軌道[26],表明W元素以W6+形態呈現。
2.1.5 紫外可見漫反射吸收光譜分析
光催化材料的光吸收范圍是評價光催化性能的重要指標。圖5(a)所示為UV?Vis DRS光譜圖,WO3單體最大的吸收波長為483nm,BiOCl0.7I0.3單體的最大的吸收波長為508nm,表明兩者均能吸收可見光。BW?15 復合材料相較于兩種單體材料最大吸收邊更寬一些,為558nm。說明BiOCl0.7I0.3和WO3兩種材料的復合拓寬了光催化劑的可見光吸收范圍,增強了可見光響應能力,能夠產生更多的活性物質,具有更高的光催化活性[27]。
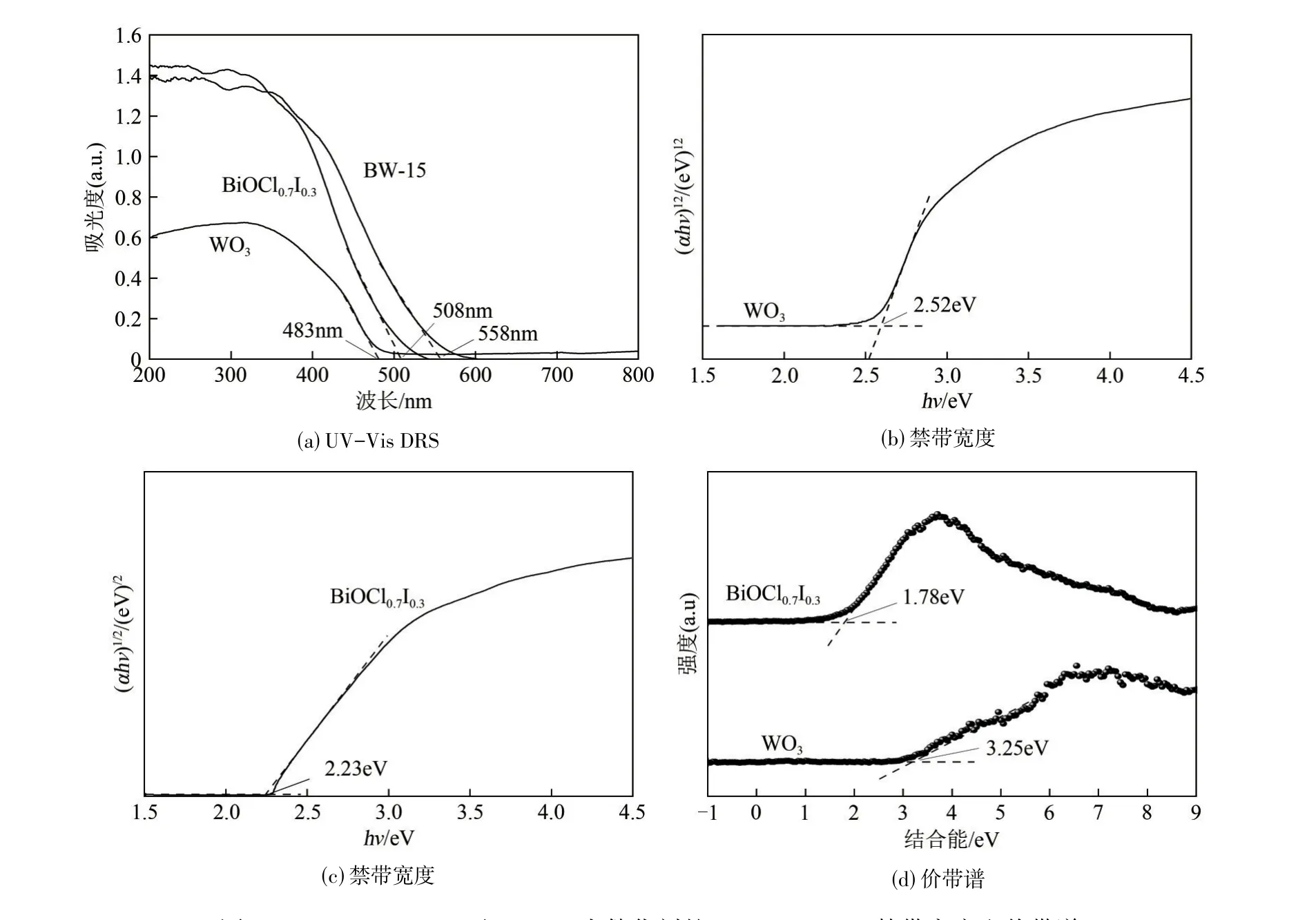
圖5 WO3、BiOCl0.7I0.3及BW?15光催化劑的UV?Vis DRS、禁帶寬度和價帶譜
BiOCl0.7I0.3和WO3均為間接帶隙半導體[27?28],其禁帶寬度(Eg)可通過式(1)計算。
式中,α、h、ν、A、Eg分別代表吸收系數、普朗克常量、光頻率、常數和禁帶寬度。
半導體的躍遷特性分為兩種,即直接躍遷和間接躍遷。其中前者n=1,后者n=4。由于BiOCl0.7I0.3和WO3均為間接躍遷的半導體,因此指數n均取4。以(αhν)1/2為縱坐標(Y)、hν為橫坐標(X)作圖,曲線的切線在X軸上的截距即為禁帶寬度,如圖5(b)、(c)所示。圖中清晰地顯示了WO3和BiOCl0.7I0.3的禁帶寬度分別為2.52eV和2.23eV。
BiOCl0.7I0.3和WO3的XPS 價帶譜如圖5(d)所示。利用XPS 價帶譜確定了BiOCl0.7I0.3的EVB=1.78eV,WO3的EVB=3.25eV。WO3和BiOCl0.7I0.3的ECB的位置可利用式(2)計算。
式中,ECB、Eg、EVB分別表示導帶電位、禁帶寬度、價帶電位。
由計算可以得出,BiOCl0.7I0.3和WO3的ECB分別為?0.45eV和+0.73eV。
2.2 光催化劑活性及穩定性
2.2.1 WO3/BiOCl0.7I0.3對鹽酸四環素的光催化降解
以鹽酸四環素為目標污染物,以不投加任何光催化劑的100mL 鹽酸四環素溶液(20mg/L)作為空白組對照,來測定所制備的WO3、BiOCl0.7I0.3以及不同配比BW?x復合光催化材料的性能。由圖6(a)可以看出,空白組的鹽酸四環素溶液濃度一直保持在比較恒定的狀態,表明鹽酸四環素具有一定的穩定性。其余幾組在經過30min的暗吸附階段后,均對鹽酸四環素有一定程度的吸附去除效果,其中BW?x復合材料的吸附能力均高于單一的WO3和BiOCl0.7I0.3,可能是因為BW?x復合材料較單體材料的比表面積有所增大,更有利于吸附污染物。在光降解階段,圖6(a)中能清晰地看到BW?x復合光催化材料對鹽酸四環素的降解能力均高于單一的WO3(34.37%)和BiOCl0.7I0.3(69.83%),其中BW?15 對鹽酸四環素的降解率最高,達到93.84%。
光催化降解鹽酸四環素符合一級反應動力學,可以利用式(3)對試驗數據進行擬合[29]。
式中,C0為目標污染物鹽酸四環素的初始濃度,mg/L;t為光照時間,min;Ct為t時刻溶液中鹽酸四環素的濃度,mg/L;k為反應速率常數。
不同光催化劑降解鹽酸四環素的偽一級動力學模型如圖6(b)所示。BW?15 的反應速率常數為0.02391min?1,遠遠大于單一的BiOCl0.7I0.3(0.00963min?1)和WO3(0.00216min?1),是BiOCl0.7I0.3的2.47 倍、WO3的11 倍。圖中清晰地顯示了BW?5、BW?10、BW?20 的k值分別為0.01205min?1、0.02164min?1、0.01998min?1,均低于BW?15,由此可以看出復合材料BW?15的光催化性能最好。
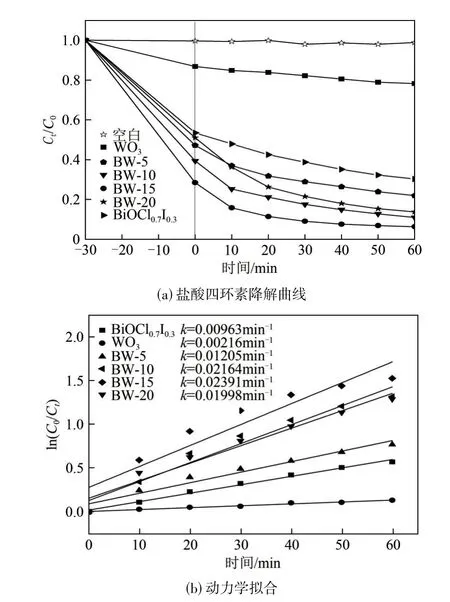
圖6 不同光催化劑對鹽酸四環素的降解及動力學擬合
由以上分析可以看出,將適量WO3與BiOCl0.7I0.3復合后能夠顯著提高光催化能力,且復合材料中組成相的比例會影響復合材料的光催化性能,WO3的含量過少會影響材料的電子轉移能力,含量過多會產生屏蔽效應[30]。
2.2.2 礦化試驗
為進一步評估所制備催化劑的光降解性能,在進行光催化降解鹽酸四環素試驗的同時進行了礦化試驗,礦化試驗采用TOC分析儀進行測定[30]。光降解1h 后,BW?15 復合材料對鹽酸四環素的礦化率為83.27%,略低于光催化降解率,如圖7所示。這是由于光降解鹽酸四環素后,溶液中部分中間產物沒有完全礦化,但相較于WO3(18.92%)和BiOCl0.7I0.3(55.64%)的礦化率依舊有顯著提高。
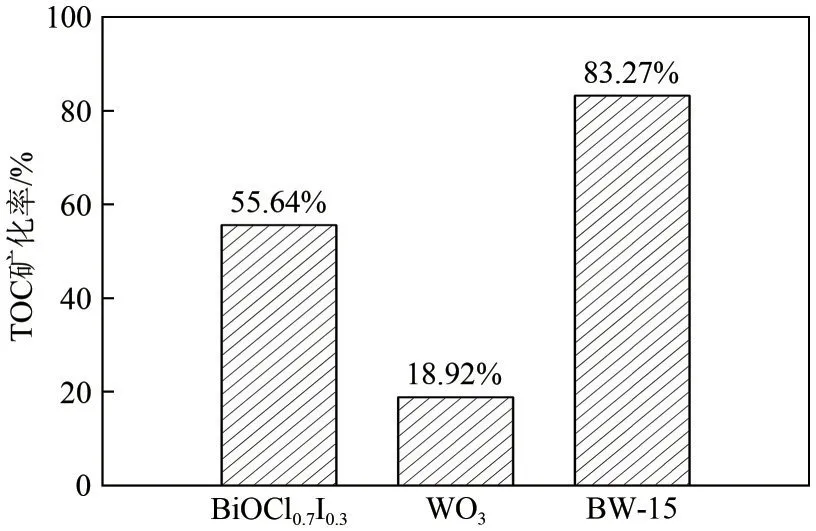
圖7 WO3、BiOCl0.7I0.3及BW?15光催化劑的礦化試驗
2.2.3 循環試驗
為了探究制備光催化劑的穩定性和可重復性,對BW?15 進行多次循環試驗。如圖8 所示,在進行了4次光降解鹽酸四環素的試驗后,BW?15的光降解率下降了13.85%,這可能是由于每次試驗后回收樣品均會有部分損耗所致。但盡管如此,4次循環試驗后,其對鹽酸四環素的去除率仍能達到79.99%,這表明BW?15 復合材料具有良好的穩定性。

圖8 BW?15復合光催化劑的循環試驗
2.3 自由基捕獲試驗
采用EDTA?2Na、IPA、BQ分別作為h+、?OH、?的捕獲劑[13],進行自由基捕獲試驗,以明確光催化過程中主要活性物質的種類,探究光催化劑降解鹽酸四環素的作用機制。以不添加光催化劑、其余條件均相同的鹽酸四環素溶液作為空白組;以加入BW?15 但不添加任何自由基捕獲劑的鹽酸四環素溶液作為對照組;以在鹽酸四環素溶液中分別加入EDTA?2Na、IPA、BQ三種自由基捕獲劑作為試驗組,進行光催化劑對鹽酸四環素的光催化降解試驗,光催化時間為60min。如圖9 所示,在目標污染物的溶液中分別加入三種不同的自由基捕獲劑后,光催化劑對鹽酸四環素的降解率均有不同程度的下降。在加入EDTA?2Na 后,降解率由93.84%降至24.50%,下降了約3.8 倍;加入BQ 后,鹽酸四環素的降解率降至56.30%,比對照組降低了37.54%;在加入IPA 后,降解率降至80.7%。因此,光催化降解過程中活性基團的影響程度為:h+>?O2?>?OH。其中h+和?O2?為該光催化反應中的主要活性物質。
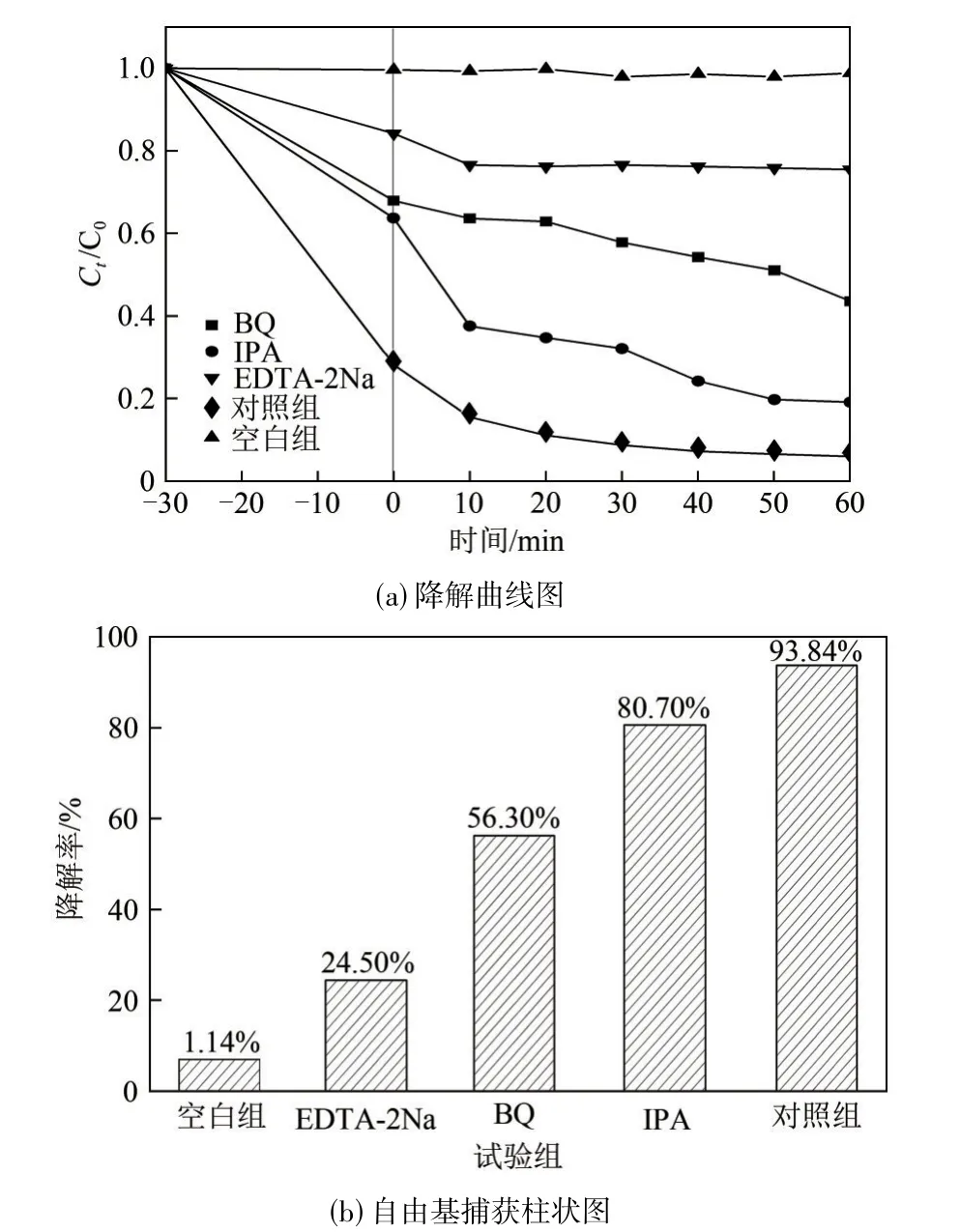
圖9 加入EDTA?2Na、IPA及BQ不同自由基捕獲劑后鹽酸四環素的降解
2.4 電子自旋共振光譜分析
光催化過程中,光照激發產生光生電子,光生電子與空穴完全分離時稱為光生載流子,參與電子轉移的氧化還原反應,?OH或?O2?是光催化氧化反應過程中的重要活性物質,光生載流子參與?OH或?的生成過程,載流子的存在與否以及濃度大小可由ESR 信號的強弱判斷[31]。?O2?的ESR 的特征強度比為1∶1∶1∶1,如圖10(a)所示;?OH 的ESR 特征強度比為1∶2∶2∶1,如圖10(b)所示。由ESR圖譜可以看出,在黑暗條件下?OH和?O2?均未產生明顯的ESR 信號,可見光照射3min 后,兩者均產生了較為明顯的特征峰信號。?O2?的信號強度較?OH 明顯更強,說明BW?15 復合材料光激發所產生的?O2?濃度高于?OH。這一結果與自由基捕獲試驗相符合。

圖10 BW?15復合光催化劑的ESR光譜
2.5 光催化機理分析
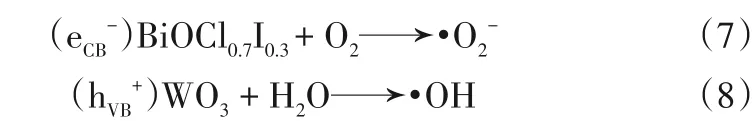
由UV?Vis DRS 以及XPS 價帶譜分析計算,可以得出制備的BiOCl0.7I0.3和WO3的導帶和價帶電位,其中BiOCl0.7I0.3的價帶電位EVB為1.78eV,導帶電位ECB為?0.45eV,導帶電位相較于O2/?O2?(?0.33eVvs. NHE)的更負,因此BiOCl0.7I0.3導帶上的光生電子可以將吸附在材料表面的O2還原為?O2?[32]。WO3的價帶電位EVB為3.25eV,導帶電位ECB為0.73eV。對于WO3來說,其價帶電位比?OH/H2O(2.4eVvs.NHE)的更正,因此WO3價帶上的空穴可以將H2O氧化為?OH[33]。
基于以上分析,WO3/BiOCl0.7I0.3復合材料可能的光催化機制如圖11 所示,WO3和BiOCl0.7I0.3構成了Z型光催化結構。可見光下,BiOCl0.7I0.3和WO3價帶上的電子受到光照激發躍遷至導帶,形成光生電子空穴對,在內部電場作用下,WO3導帶上的電子轉移至BiOCl0.7I0.3價帶上,同其上的光生電子一樣躍遷至BiOCl0.7I0.3的導帶上,并在其上累積。反應式見式(4)~式(6)。

圖11 WO3/BiOCl0.7I0.3復合光催化材料的光催化機制
此時,由于BiOCl0.7I0.3和WO3產生的光生電子都累積在BiOCl0.7I0.3的導帶上,有效地分離了電子空穴對,因而提高了材料的光催化活性。BiOCl0.7I0.3導帶電位為?0.45eV,比O2/?O2?(?0.33eVvs. NHE)的更負,可以將材料表面吸附的O2還原為?O2?并參與鹽酸四環素的降解,這也讓電子可以快速清除,使得光催化劑可以產生更多的活性基團。與此同時,WO3的價帶電位EVB為3.25eV,比?OH/H2O(2.4eVvs. NHE)的更正,因此WO3價帶上由于電子躍遷形成的空穴一部分直接參與光催化反應,另一部分將H2O 氧化為?OH 再參與反應,而BiOCl0.7I0.3價帶上的空穴將直接參與光催化降解反應。這與2.3 節中自由基捕獲試驗結果相符合。反應式見式(7)、式(8)。
h+/?O2?/?OH + 鹽酸四環素→降解產物(CO2+ H2O)(9)
3 結論
(1)采用煅燒法、水浴加熱和原位沉淀法成功制備了WO3/BiOCl0.7I0.3復合光催化材料。通過XRD、SEM、FTIR、XPS 和UV?Vis DRS 等表征手段對材料進行表征分析,驗證了復合材料的成功合成。
(2) WO3/BiOCl0.7I0.3最 佳 摻 雜 比 例 為WO3∶BiOCl0.7I0.3=1∶15(摩爾比),對鹽酸四環素的光催化降解效果最優。
(3)通過光降解試驗以及循環試驗,證明WO3/BiOCl0.7I0.3對鹽酸四環素具有良好的光降解能力和穩定性,循環4 次后仍具有較高的光催化性能,表明了該材料具有可循環性。
(4)通過自由基捕獲試驗和ESR 圖譜分析,得出h+和?O2?是光催化過程中主要的活性物質,結合材料禁帶寬度和價帶譜,分析出WO3/BiOCl0.7I0.3光催化材料Z型異質結構的光催化作用機理。
猜你喜歡
建材發展導向(2022年2期)2022-03-08 01:44:04
建材發展導向(2021年14期)2021-08-23 00:56:16
中國材料進展(2019年10期)2019-12-07 05:32:14
纖維復合材料(2018年3期)2018-04-25 07:22:58
電子測試(2017年11期)2017-12-15 08:57:13
山東工業技術(2016年15期)2016-12-01 05:31:34
中國塑料(2015年6期)2015-11-13 03:02:54
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年8期)2015-10-14 01:10:41
應用化工(2014年10期)2014-08-16 13:11:29
