癌癥治療中PI3K/AKT/mTOR 通路及靶向抑制劑研究進展*
2023-03-12 14:20:02周慧海廣范張濤鄧智建
中國藥業 2023年5期
關鍵詞:乳腺癌
周慧,海廣范,張濤,鄧智建
(新鄉醫學院藥學院,河南 新鄉 453003)
國際癌癥研究中心2020年發布的《世界癌癥報告》統計顯示,全世界179個國家中癌癥是導致過早死亡的重要因素,發病率逐年上升[1]。分子靶向治療作為腫瘤治療的重要方法,是以病變組織、細胞中的特殊分子作為靶點,特異性抑制癌細胞生長增殖。磷脂酰肌醇3 激酶/ 蛋白激酶B/ 哺乳動物雷帕霉素靶蛋白(PI3K/AKT/mTOR)通路可被多種生長因子激活,其異常激活認為是癌癥發生的重要特征[2]。Caris 生命科學公司的一項研究發現,PI3K/AKT 通路高頻上調[3]。另一項泛癌研究對癌癥基因組圖譜(TCGA)數據庫的32 種11 219 個腫瘤樣本分析發現,PI3K/ AKT 通路異常上調[4]。PI3K/AKT/mTOR通路的上調對抑制凋亡基因表達、誘導保護性自噬、加速新生血管形成等具有重要作用,目前此通路在癌癥靶向治療中的作用已受到密切關注。在此,歸納PI3K/AKT/mTOR通路在多類型癌癥進展中的作用,比較該通路靶向抑制劑的治療作用,以為PI3K/AKT/mTOR通路抑制劑的開發臨床應用提供參考。
1 對癌癥進展的影響
1.1 抑制癌細胞凋亡,誘導腫瘤形成
PI3K/AKT/mTOR 信號通路可調控一系列凋亡相關因子,包括Bcl-2 基因家族、X-連鎖凋亡抑制蛋白(XIAP)、鼠雙微基因2(MDM-2)、Forkhead box O3 轉錄因子(FOXO3a)等。AKT 磷酸化促凋亡蛋白B 淋巴細胞瘤-2基因相關啟動子(BAD),使其失去結合Bcl-xL形成異源二聚體的能力,導致Bcl-xL 蛋白從二聚體中釋放,發揮抗凋亡作用[5]。AKT 還可磷酸化MDM-2 的Ser166 和Ser186 位點,使MDM-2 結合并阻斷腫瘤蛋白p53 基因(TP53)N 端反式激活結構域,導致p53 蛋白泛素化后降解[5]。p53 可促進凋亡蛋白Bcl-2 相關X 蛋白(BAX)表達,當p53缺失時促生存蛋白Bcl-2與BAX間的平衡被破壞,引起細胞抵抗凋亡。XIAP是一種細胞內抗凋亡蛋白,具有3個桿狀病毒細胞凋亡抑制蛋白(IAP)重復序列(BIR)結構域,可被AKT 磷酸化激活,激活的XIAP的BIR結構域可直接結合Caspase家族,抑制促凋亡蛋白caspase 3,caspase 8,caspase 9發揮作用[6]。
ZHOU 等[7]的研究證實,丹參酮Ⅰ對卵巢癌的抑制作用是通過下調PI3K,AKT,mTOR 蛋白磷酸化,抑制PI3K/ AKT/ mTOR 通路激活而產生的,該信號通路的下調進一步促進了caspase 3,cleaved caspase 3 的表達上升,Bcl-2/BAX 比例下降;在小鼠體內實驗中,藥物處理組小鼠腫瘤體積、質量顯著小于對照組,且腫瘤組織蛋白檢測結果驗證了丹參酮Ⅰ的抗腫瘤作用與PI3K/AKT/ mTOR 通路下調的密切關系。另一項敲低Inc RNA CASC9 的實驗中發現,抑制口腔鱗狀細胞癌細胞AKT/ mTOR 通路激活后,Bcl-2/BAX 比例顯著下降,腫瘤細胞增殖能力降低、凋亡增多;在小鼠體內移植瘤實驗中,小鼠腫瘤體積、質量均顯著小于對照組[8]。關于肝癌[9]、胃癌[10]、甲狀腺癌[11]的研究證明,下調PI3K/AKT/mTOR通路激活對癌細胞增殖、生長具有抑制作用。
1.2 對癌細胞遷移、侵襲的影響
PI3K/ AKT/ mTOR 信號通路激活可促進癌細胞的遷移。激活后的AKT 誘導mTORC1 及其下游靶點如4E-BP1和p70S6K發生磷酸化,導致上皮間質轉化(EMT)標記物波形蛋白和神經細胞鈣黏蛋白(N-cadherin)的上調及上皮標記物上皮細胞鈣黏蛋白(E-cadherin)、緊密連接蛋白1(ZO-1)、連接蛋白(claudin)的減少[12],可使細胞的轉移、侵襲能力上升。在胃癌組織中,N6-腺苷酸甲基化(m6A)的mRNA 水平低于臨近正常組織樣本。LIU 等[13]發現,過表達甲基轉移酶樣蛋白14抗體(METTL14)可抑制PI3K/ AKT/ mTOR 通路激活,對基質金屬蛋白酶9(MMP9)、E-cadherin、N-cadherin等蛋白的表達具有下調作用,故認為METTL14 對胃癌細胞侵襲的抑制作用可能與抑制PI3K/AKT/mTOR 的激活有關。
活化的mTORC1 通過激活p70 核糖體蛋白S6 激酶(p70S6K)調節Rac1 和Cdc42,控制癌細胞運動過程中的肌動蛋白重組,影響癌細胞的遷移。p70S6K激活后還可直接與交聯的F-肌動蛋白(F-actin)相互作用,抑制絲切蛋白(cofilin)家族肌動蛋白解聚。基質金屬蛋白酶(MMP)在細胞侵襲中可降解細胞外基質,p70S6K 可促進MMP 的激活與表達[14],使細胞更易發生侵襲。木犀草素抑制三陰乳腺癌轉移的機制研究結果顯示,經木犀草素處理的人乳腺癌細胞(BT-20 細胞)的AKT和mTOR 蛋白磷酸化水平顯著降低,導致基質金屬酶2(MPP-2)和MPP-9 mRNA、蛋白水平下調;體外細胞遷移實驗和劃痕實驗中,木犀草素對過表達豆蔻酰化AKT(myr-AKT)的BT-20 細胞幾乎無作用[15],表明通過抑制AKT/ mTOR 磷酸化激活,下調MPP 家族表達,對癌細胞侵襲具有抑制作用。
1.3 對癌細胞化療耐藥的影響
自噬是癌細胞產生化療耐藥的重要因素。細胞通過自噬避免在不利生長環境中受損。自噬作為一種程序性死亡過程,過度自噬通過降解細胞內異常蛋白、受損細胞器,引起細胞死亡[16]。mTOR 作為PI3K/ AKT/mTOR 通路的重要成分,抑制該通路激活,影響下游mTOR 誘導的自噬,對降低癌癥化療耐藥具有重要作用。YANG 等[17]的研究發現,芹菜素對肝癌細胞具有凋亡誘導作用,芹菜素僅能減小雄性移植瘤模型小鼠腫瘤體積,移植瘤質量較對照組無顯著變化。3-甲基腺嘌呤(3-MA)作為一種Ⅲ類PI3K 抑制劑[18],與芹菜素聯用后可抑制PI3K/AKT/mTOR 通路激活,導致自噬標志物微管相關蛋白1輕鏈3B-Ⅱ(LC3B-Ⅱ)蛋白表達水平顯著降低,移植瘤體積、質量較芹菜素治療組下降[17],表明PI3K/AKT/mTOR 通路抑制對提高抑癌藥物療效,增加腫瘤敏感性具有顯著作用。
除了通過抑制PI3K對整個通路磷酸化激活進行下調,直接抑制mTOR 或雙靶向抑制PI3K/mTOR 對治療癌癥自噬性耐藥具有積極作用。DENG 等[19]研究發現,在耐藥上皮性卵巢癌(EOC)細胞中,與單用順鉑處理組相比,PI3K/ mTOR 抑制劑BEZ235 與順鉑聯用處理腫瘤細胞可顯著抑制克隆團形成,促進細胞凋亡,逆轉上皮間質轉化(EMT)發生,下調化療耐藥標志物癌癥干細胞(CSC)表達,使耐藥細胞EOC 重新敏感。另一項BEZ235 治療阿霉素耐藥的K562 細胞研究[20]證實,抑制PI3K/AKT/mTOR 通路激活有利于使耐藥細胞重新敏感,增強化療效果。
由圖1 可知,PI3K/ AKT/ mTOR 通路通過磷酸化作用逐級激活,可調節影響細胞生長、增殖、遷移、侵襲及凋亡的多種因子,以及促進腫瘤進展的多個環節,PI3K/AKT/mTOR 通路復雜多樣的生理功能為該通路抑制劑的開發應用提供了堅實的基礎。
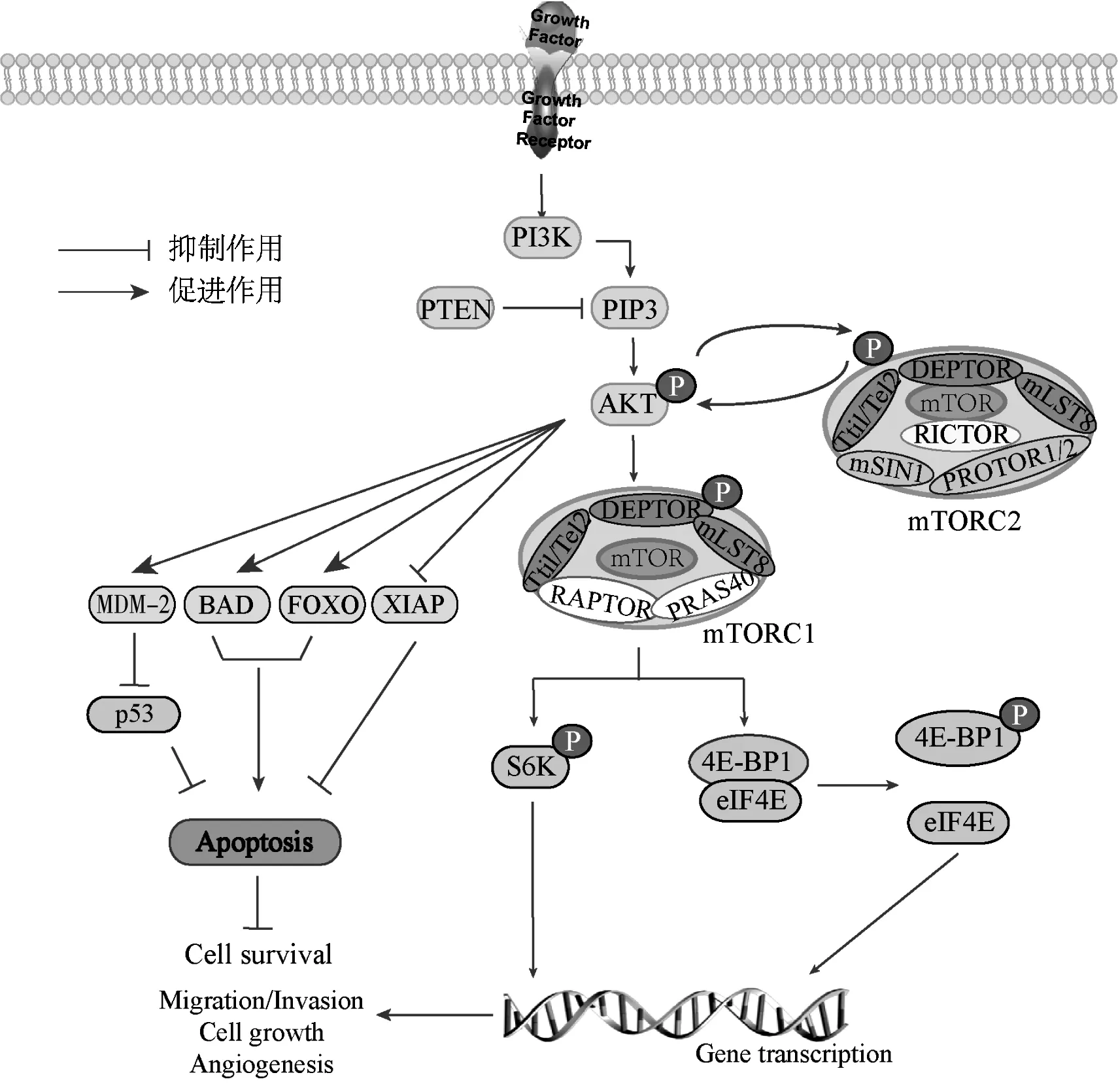
圖1 PI3K/AKT/mTOR 通路在細胞中的信號傳導與作用Fig.1 Signal transduction and roles of PI3K/AKT/mTOR pathway in cells
2 靶向抑制劑及其作用
2.1 PI3K 靶向抑制劑
2.1.1 廣譜PI3K 抑制劑
新型廣譜PI3K 抑制劑具有耐受性好、不良反應少等優點,與其他治療方法或臨床抑癌藥物配合治療效果良好。目前,常見廣譜PI3K 抑制劑包括LY294002,BKM120(buparlisib),copanlisib,PX-866,GDC-0941(pictilisib),PX-866,XL-147,GNE-317等。
LY294002 作為第1 個廣譜PI3K 抑制劑,對多種腫瘤細胞的PI3K 相關通路具有廣泛抑制作用,臨床研究中常與阿霉素、長春新堿、5-氟尿嘧啶、依托泊苷等抑癌藥物聯用,可提高藥物療效、減緩耐藥性產生[21],但未見單藥抗腫瘤作用評價的報道。
BKM120 可廣泛抑制PI3K 的p110 α,β,δ,γ 亞型,但對p110 α 亞型的抑制效果更好,能以ATP 競爭方式抑制次級信使磷脂酰肌醇-3,4,5-三磷酸(PIP3)激活。ZHAO 等[22]評估353 個腫瘤細胞系對BKM120 的作用發現,BKM120 對PI3KCA 突變的腫瘤細胞系具有優先抑制作用,而PI3KCA 突變與乳腺癌進展又密切相關。在BKM120 治療PI3K 通路激活的晚期乳腺癌的Ⅲ期臨床試驗中,BKM120 治療組患者無進展生存期中位數為6.8 個月,顯著長于安慰劑組的4.0 個月[23],表明其可延長患者的生存期。
Copanlisib 是一種泛Ⅰ型PI3K 抑制劑,主要作用于PI3Kα 和δ 亞型,均對促進B 淋巴細胞存活和遷移至骨髓方面具有重要作用[24]。2017年,copanlisib 由美國食品和藥物管理局(FDA)批準用于治療難治性或復發性濾泡性淋巴瘤[24];在霍奇金病的Ⅲ期臨床試驗中,與利妥昔單抗聯用顯著延長患者的無進展生存期[25]。
泛PI3K抑制劑對各種實體瘤均展現出一定抑制作用,但非選擇性阻斷PI3K 亞型導致大量不良反應。BKM120 對乳腺癌的Ⅱ期臨床研究中,12%的患者臨床獲益,但高血糖、厭食癥等三級及以下不良反應發生率超30%[26];日本一項廣譜抑制劑GDC-0941(pictilisib)治療晚期實體瘤患者的Ⅰ期臨床試驗中,給藥340 mg/d后出現3 級黃斑丘疹[27];一項GDC-0941(pictilisib)聯合常見抗癌藥物治療晚期乳腺癌的Ⅰb 期試驗中,患者出現不同程度中性粒細胞減少(44.9%)、皮疹(50.7%)、腹瀉(78.3%)等[28]。部分泛PI3K 抑制劑在臨床階段未達到臨床前研究中展現出的抗腫瘤作用,導致可用于臨床的泛PI3K抑制劑十分有限。
2.1.2 選擇性PI3K 抑制劑
在腫瘤發生過程中,部分PI3K亞型常發生突變,進行靶向抑制可減少不良反應,提高藥物療效。其中,PI3Kα亞型選擇性抑制劑對PI3KCA突變的惡性腫瘤具有更好的抑制作用[29]。VASAN 等[30]研究報道了順式PI3KCA 雙突變與乳腺癌發病的密切關系,15%的患者的病變組織中存在PI3K 異常激活,且可被PI3K 抑制劑有效控制,對PI3Kα 抑制劑更加敏感。PI3Kα 亞型抑制劑BKM120[31]和GDC-09411[32]及選擇性PI3Kα 抑制劑alpelisib等對乳腺癌具有一定的治療作用。
BYL719(alpelisib)是選擇性PI3Kα 抑制劑。體內實驗中,alpelisib 以時間、劑量依賴方式抑制PI3K 信號通路激活,表現出較強的抗腫瘤作用,具有良好的耐受性。磷脂酰肌醇3激酶催化亞基a基因(PI3KCA)認為是alpelisib 敏感的重要生物標志物[33]。在治療乳腺癌方面,alpelisib 對絕經后HR 陽性/人表皮生長因子受體2(HER2)陰性且攜帶PI3K突變的晚期患者治療作用良好。在Ⅲ期臨床試驗中,相比于安慰劑,BYL719(alpelisib)聯合氟維司群可有效延長患者無進展生存期[34]。2019 年5月,FDA 批準BYL719(alpelisib)聯合氟維司群用于治療男性或絕經后女性HR 陽性/HER2 陰性且攜帶PI3K突變的晚期乳腺癌[35]。
亞型特異性PI3K抑制劑的毒性往往取決于其抑制的具體亞型,PI3Kα選擇性抑制劑通常會引起高血糖或皮疹,除了BYL719(alpelisib)外,TAK-117(serabelisib)作為口服選擇性PI3Kα抑制劑同樣具有上述不良反應。serabelisib(TAK-117)的Ⅰ期臨床研究發現,其在晚期實體瘤中具有單藥抗腫瘤活性,但藥物相關Ⅲ級不良反應發生率為27%,主要不良反應有高血糖(8%)、3 級丙氨酸氨基轉移酶/ 天門冬氨酸氨基轉移酶(ALT/AST)升高(15%)等,其藥物安全性不足,限制了臨床的進一步臨床使用[36]。
Idelalisib是一種口服ATP結合位點競爭性抑制劑,對PI3Kδ選擇性高于其他PI3K亞型30倍以上[37]。2014年7月,FDA 批準idelalisib 用于復發性、難治性慢性淋巴細胞白血病的治療。Idelalisib與美羅華聯用可有效提高患者的客觀緩解率、總生存期[38],表現出良好的治療作用,常見輕度或中度不良反應。PI3Kδ 抑制劑常伴有胃腸道反應、骨髓抑制、轉氨酶升高等[39],對PI3Kδ亞型具有較高選擇性的抑制劑idelalisib,duvelisib,umbralisib,copanlisib,acalisib,parcaclisib,ME-401。治療慢性淋巴細胞白血病(CLL)時,采用間斷給藥或減量可降低不良反應發生率[31]。
多種PI3K抑制劑已進入不同的臨床發展階段,已獲FDA批準用于臨床的PI3K抑制劑仍十分短缺。目前,僅有用于血液系統惡性腫瘤的PI3K 抑制劑idelalisib 和copanlisib,以及用于乳腺癌的選擇性抑制劑alpelisib。
2.2 AKT 靶向抑制劑
2.2.1 概述
AKT 可分為AKT1,AKT2,AKT3 3 種亞型。目前,在胃癌、卵巢癌、胰腺癌等多種惡性腫瘤中均發現了3 種亞型的異常擴增,在部分惡性腫瘤如乳腺癌、結腸癌、肝細胞癌及黑色素瘤中還存在AKT 亞型的過表達現象[40],靶向抑制AKT已成為癌癥治療的思路之一。
2.2.2 ATP 競爭性AKT 抑制劑
ATP 競爭性抑制劑可直接靶向具有催化活性的激酶結構域,從而抑制AKT[41]。Ipatasertib(GDC-0068)是一種ATP 競爭性、選擇性泛AKT 抑制劑,口服對AKT的3 種亞型均有良好的抑制作用,對蛋白激酶A(PKA)家族的其他成員抑制作用較差,具有較好的選擇性。在腫瘤細胞及腫瘤異種移植模型中,經ipatasertib 處理的腫瘤細胞周期進展被阻滯,細胞的生存能力降低,在AKT活化水平增高、類脂磷酸酶(PTEN)缺失及PI3KCA突變的情況下均有良好的腫瘤細胞抑制作用[42]。Ⅰ期臨床試驗表明,ipatasertib 耐受性、吸收性良好[43];單藥抗腫瘤Ⅰ期臨床試驗中,ipatasertib 對30%實體腫瘤患者表現出抗腫瘤活性,包括結腸癌、卵巢癌、前列腺癌等,8.5%的患者無進展生存期超過6個月,不良反應多為1~2 級胃腸道反應,部分患者出現高血糖(10%)、皮疹(6%)等[39]。Ipatasertib 與阿比特龍+潑尼松龍[44]或阿比特龍[45]聯用治療前列腺癌的一項Ⅲ/Ⅱ期臨床試驗中,可有效延長PTEN 缺失的前列腺癌患者的無進展生存期,表現出良好的治療作用。
GSK2141795 或GSK795,又稱uprosertib,是一種口服ATP 競爭性AKT 抑制劑。GSK2141795 在多種腫瘤細胞系中可抑制AKT 下游底物磷酸化,產生有效的抗腫瘤活性,但AKT自身磷酸化水平在體內外均升高[46]。相比于未激活的AKT,ATP 競爭性抑制劑與激活的AKT親和性更高。通過將AKT 鎖定在磷酸化但無功能狀態,阻斷AKT 激活下游信號通路,達到抑制腫瘤細胞的目的。AGHAJANIAN 等[47]研究發現,在GSK2141795 抗腫瘤作用Ⅱ期臨床研究中,GSK2141795 對PTEN 缺失、PI3KCA突變實體瘤患者療效更好。
GSK-690693 作為第1 個進入臨床試驗的APT 競爭性抑制劑,由于口服生物利用度低,且不良反應嚴重,阻礙了Ⅰ期臨床試驗的研究。LY-2780301 是一種p70S6K 與AKT 的雙重抑制劑,在Ⅰ期臨床試驗中出現多種不良反應,包括便秘(19%)、疲乏(13%)、惡心(9%)、腹瀉(9%)[48],但與紫杉醇聯用治療激素耐藥HER2陰性或三陰性晚期乳腺癌對PI3K/AKT激活患者的客觀緩解率達63.9%,展現出良好的抗腫瘤作用[49]。
對比ipatasertib,LY-2780301,GSK2141795 臨床研究結果發現,僅抑制AKT 下游底物磷酸化的抗腫瘤作用弱于直接靶向AKT,故ipatasertib 的發展前景相對優于GSK2141795和LY-2780301。
2.2.3 AKT 變構抑制劑
變構激酶抑制劑是目前研究AKT 抑制劑的方向之一,其結合和作用區域都在ATP 結合位點外。AKT 的3 種異構體都包含PH 結合域,其中的色氨酸殘基(Trp80)吸引變構抑制劑與其結合并產生相互作用[50],在膜水平阻斷AKT 與磷脂酰肌醇-3,4,5-三磷酸(PIP3)的結合,抑制AKT的活化。
MK-2206是一種口服變構型AKT 抑制劑,可靶向抑制AKT的3種亞型。AKT磷酸化水平在順鉑耐藥細胞系(AGS,MGS-803,MKN-45)中較高,與單用順鉑治療相比,順鉑聯用MK-2206可使凋亡標志物二磷酸核糖聚合酶(PARP)表達增加,對癌細胞產生更強的抑制作用[51]。MK-2206治療淋巴瘤的Ⅱ期臨床試驗中,56例患者完全緩解2例,經典霍奇金淋巴瘤25 例患者有效率為20%,Ⅲ級不良反應(15%)[52]。在另一項Ⅱ期臨床試驗中對比MK-2206 與依維莫司對難治性腎癌的作用,按2∶1 將43 例患者隨機分為MK-2206 組、依維莫司組,結果顯示,MK-2206 無進展生存期弱于依維莫司,但MK-2206治療組患者治療效果明顯優于依維莫司組,MK-2206 誘導皮疹、瘙癢等不良反應發生率明顯高于依維莫司組[53]。
2.3 mTOR 靶向抑制劑
2.3.1 雷帕霉素及其衍生物
抗生素變構mTOR 抑制劑主要包括雷帕霉素及其衍生物(rapalogs),可靶向抑制mTOR 和FKBP12,作用基本一致。Rapalogs與FKBP12結合形成復合物,復合物結合mTOR 的FRB 結構域,改變mTOR 的構象,抑制mTORC1激酶的活性[54]。
雷帕霉素最初被認為是一種具有抗真菌、免疫抑制、抗癌細胞增殖的藥物。后續研究發現,雷帕霉素通過與FKBP12 蛋白結合抑制mTORC1 發揮藥理作用,但雷帕霉素溶解性、藥代動力學差,不適合用于人類癌癥的治療[25]。目前,雷帕霉素類似物有依維莫司、temsirolimus、ridaforolimus。依維莫司經FDA批準用于ER陽性/HER 陰性乳腺癌治療口服雷帕霉素類似物。在治療晚期非功能性肺或胃腸道神經內分泌腫瘤的Ⅲ期臨床研究中,依維莫司可使疾病進展或死亡的估計風險降低52%[55]。在依維莫司聯合來曲唑與來曲唑單藥治療絕經前HR 陽性/ ERBB2 陰性的晚期乳腺癌的Ⅱ期臨床試驗中,依維莫司聯合來曲唑組的無進展生存期明顯長于單獨服用來曲唑組,依維莫司有效增加了來曲唑的抗腫瘤作用[56]。
相比于雷帕霉素,temsirolimus(torisel)水溶性與穩定性更好[57]。Temsirolimus 通過細胞色素P4503A4 酶(CYP3A4)轉化為雷帕霉素而發揮作用,可維持數天[31]。因此,使用temsirolimus 治療過程中應密切監控CYP3A4 指標,及時調整藥物用量。Temsirolimus 在治療復發性/ 難治性原發性中樞神經系統淋巴瘤(PCNSL)的Ⅱ期臨床研究中,患者總緩解率達54%,常見不良反應為高血糖(29.7%)、血小板減少(21.6%)、感染(19%)等。收集患者腦脊液,75 mg 靜脈滴注組中僅1 例檢測到temsirolimus,其余患者腦脊液中未發現藥物,說明單用temsirolimus 治療復發性/難治性PCNSL 具有抗腫瘤活性,但作用短暫[58]。
Ridaforolimus(AP23573,MK-8669)是一種雷帕霉素的非前藥類似物,對mTOR 具有特異性抑制作用[59]。有研究顯示,ridaforolimus 與達洛妥珠單抗聯用對雌激素受體陽性乳腺癌具有一定的抗腫瘤活性[60]。另一項ridaforolimus 與紫杉醇、卡鉑聯用治療實體瘤的Ⅰ期臨床試驗中,50%的患者部分緩解,33%的患者病情穩定,18 例患者實體瘤評估中腫瘤體積下降25%[61],對抑制腫瘤生長及延緩病情發展具有良好作用。
常見的mTOR 抑制劑依維莫司、temsirolimus 已被FDA 批準分別用于腎細胞癌、晚期腎細胞癌的治療。臨床研究發現,雷帕霉素衍生物的單藥抗腫瘤作用有限,但與其他抗癌藥物聯用時可有效提高療效。
2.3.2 ATP 競爭性mTOR 靶向抑制劑
ATP競爭性mTOR抑制劑及mTOR/PI3K雙抑制劑為小分子的ATP類似物,可直接作用于mTOR或PI3K的ATP 結合位點,產生競爭性抑制作用,不僅能抑制細胞生長,還可誘導腫瘤細胞凋亡。ATP競爭性mTOR抑制劑對mTORC1和mTORC2均可產生抑制作用。
MLN0128(INK128,TAK228)是一種泛mTOR 抑制劑,在體內外都有抗腫瘤作用。MLN0128 能抑制CD44高表達的腫瘤外植體的生長,且能使耐藥的肝細胞癌對索拉非尼重新敏感[62]。
LY3023414 是一種PI3K,mTOR,DNA-PK 的ATP競爭性抑制劑,Ⅰ期臨床研究中,患者發生的主要不良反應包括惡心(38%)、疲勞(34%)、嘔吐(32%)等,大多數為輕度或中度,藥物劑量≥150 mg 時靶向抑制率≥90%,表明LY3023414對于晚期癌癥患者具有可耐受的安全性及單藥活性[63]。
ATP 競爭性mTOR 抑制劑通過抑制mTOR 而有效降低由單一抑制mTORC1 產生的上游mTORC2 的反饋激活,對減緩耐藥性產生,提高藥物療效具有積極作用。與雷帕霉素類似物相比,部分ATP 競爭性mTOR 抑制劑的單藥抗腫瘤活性相對更好。
2.3.3 其他mTOR 抑制劑及天然活性產物
由于mTOR 是以mTORC1 和mTORC2 形式存在并發揮作用,因此抑制亞基間相互作用可對mTOR產生抑制作用。P529 可識別Rictor 和Raptor,并發揮相互作用,抑制mTORC2 和mTORC1 復合物形成,JR-AB011 可識別結合Rictor,從而發揮抑制mTORC2的作用。
一些天然產物對mTOR 及其所在通路具有抑制作用,如在姜黃、菖蒲中分離提取的curcumin 具有抗炎、抗增殖、抗侵襲、放射增敏及化學增敏等作用,能以劑量依賴方式降低mTOR,Rictor,Raptor,是一種mTORC1和mTORC2 的泛mTOR 抑制劑。NELSON 等[64]研究現,curcumin在治療包括結腸癌、胰腺癌在內的多種疾病過程中,測試對象血清中curcumin 的含量雖然較低,但低劑量下具有一定療效,中高劑量療效尚不明確。因curcumin量效關系不穩定、構效關系不明等因素制約其進一步開發利用。mTOR 抑制劑雖有一定的抗腫瘤活性,但單藥治療作用不顯著,常與其他抗腫瘤藥物聯用以提高治療作用,mTOR 抑制劑在對抗耐藥方面發揮了重要作用。
2.4 PI3K/mTOR 雙靶向抑制劑
PI3K與mTOR分別作為PI3K/AKT/mTOR 通路的兩端,靶向PI3K 有利于對提高藥物的抗腫瘤活性,而mTOR 作為多種實體瘤耐藥的關鍵蛋白,靶向mTOR 有利于產生協同抗腫瘤作用,減緩耐藥產生。PI3K/mTOR雙靶向抑制劑表現出了良好的抗腫瘤效果。
PKI-587(gedatolisib,PF-05212384)是一種泛Ⅰ類PI3K/ mTOR 抑制劑[65]。PKI-587 不僅對慢生長細胞系BON,QGP-1,KRJI 具有增殖抑制作用,對于低分化水平的快速增長細胞LCC-18 也具有抑制作用[66]。與依維莫司相比,PKI-587 處理的胃腸胰腺神經內分泌腫瘤細胞系BON,QGP-1,KRJI,LCC-18 抑制率為61%~94%,高于依維莫司處理組抑制率的47%~52%[66]。在PKI-587 治療復發性子宮內膜癌患者的Ⅱ期臨床研究中,3~4 級不良事件的發生率低于其他PI3K,mTOR 抑制劑[67]。另一項臨床研究[68]發現,PKI-587的主要不良反應為黏膜炎癥(43%)、惡心(41%)、高血糖(26%)、嘔吐(24%),未出現Ⅳ級及以上不良事件。可見,PKI-587對腫瘤細胞具有廣譜高效的抑制作用。
PQR309 作為一種口服生物可利用的PI3K/mTOR雙靶向抑制劑,對Ⅰ型PI3K具有廣泛抑制作用,同時以ATP 競爭的方式抑制mTOR 的作用,避免由于mTORC1抑制導致的mTORC2 反饋激活,對減少耐藥的產生具有積極意義。在評價PQR309的安全性、有效性的Ⅱ期臨床試驗[69]中,復發或難治性淋巴瘤患者總緩解率為14%,36%的患者病情穩定,但治療過程中70%的患者出現Ⅲ級不良反應,部分患者出現Ⅳ級不良反應,如高血糖(24%)、中性粒細胞減少(20%)、血小板減少(22%)。
SAR245409 是一種口服有效的Ⅰ型PI3Ks/ mTOR雙抑制劑,Ⅰ期臨床研究發現,SAR245409 治療復發或難治性淋巴瘤患者的最大耐受劑量(MTD)為50 mg,每日2 次[70]。Ⅱ期臨床研究發現,SAR245409 治療164 例難治性非霍奇金淋巴瘤、CLL、小淋巴細胞淋巴瘤,緩解30例,完全緩解8例。
相較于mTOR 和PI3K 單靶向抑制劑而言,PI3K/mTOR 雙靶向抑制劑有效解決了mTOR 單靶向抑制劑療效不佳的問題,客觀緩解率升高,但不良反應也增加了。亞型選擇性不僅會引發PI3Kδ 抑制,而且會發生多種不良反應,故在對PI3K/ mTOR 進行雙重抑制時,亞型選擇性是開發安全高效靶向抑制劑的有效途徑。
3 展望
PI3K/ AKT/ mTOR 信號通路是多種腫瘤發生、進展中的常見通路,為癌癥治療提供了多個靶點,PI3K/AKT/ mTOR 抑制劑主要在乳腺癌、B 淋巴細胞惡性腫瘤方面具有廣泛的抑制作用。目前,部分PI3K 抑制劑、mTOR 抑制劑已獲FDA 批準用于乳腺癌的臨床治療,PI3K,AKT,mTOR,PI3K/ mTOR 雙靶向抑制劑均已進入B淋巴細胞惡性腫瘤臨床研究階段。
PI3K,AKT,mTOR,PI3K/ mTOR 雙靶向抑制劑在抗腫瘤治療中均存在不同的作用特點。PI3KCA 突變與乳腺癌發病存在密切關系,研究多集中于乳腺癌領域,PI3KCA 突變對PI3Kα 選擇性抑制劑更敏感[30],目前選擇性PI3Kα 抑制劑alpelisib 已批準用于晚期乳腺癌的臨床治療,泛抑制劑BKM120,pictilisib 等由于其不良反應較多,難以進入下一步研究。相較于PI3K 抑制劑,AKT 抑制劑多為泛抑制劑,以ATP 競爭、AKT 變構方式實現AKT1,AKT2,AKT3 3 種亞型的抑制,這導致AKT 抑制劑不良反應多,難以進入臨床研究。但AKT抑制劑對PTEN 突變、缺失的惡性腫瘤仍有良好的發展潛力。mTOR 作為整個通路的下游,調控著細胞自噬的發生與發展程度[71],與癌癥化療耐藥的產生密切相關,在減緩其他抗癌藥物耐藥性產生,提高抗腫瘤活性方面具有重要作用,但也存在單藥抗腫瘤活性弱的缺點。
猜你喜歡
中老年保健(2022年6期)2022-08-19 01:41:48
現代臨床醫學(2022年1期)2022-02-12 02:04:58
甘肅科技(2020年20期)2020-04-13 00:30:42
中國生殖健康(2019年2期)2019-08-23 08:11:42
中國生殖健康(2019年6期)2019-01-06 09:20:12
中國生殖健康(2019年5期)2019-01-06 09:16:40
幸福家庭(2019年14期)2019-01-06 09:15:38
祝您健康(2018年5期)2018-05-16 17:10:16
癌癥進展(2016年9期)2016-08-22 11:33:20
中國組織化學與細胞化學雜志(2016年4期)2016-02-27 11:16:08
